
We conducted a genome-wide association study (GWAS) to identify novel predisposition alleles associated with Philadelphia chromosome-negative myeloproliferative neoplasms (MPNs) and JAK2 V617F clonal hematopoiesis in the general population.
We recruited a web-based cohort of 726 individuals with polycythemia vera, essential thrombocythemia, and myelofibrosis and 252 637 population controls unselected for hematologic phenotypes. Using a single-nucleotide polymorphism (SNP) array platform with custom probes for the JAK2 V617F mutation (V617F), we identified 497 individuals (0.2%) among the population controls who were V617F carriers. We performed a combined GWAS of the MPN cases plus V617F carriers in the control population (n = 1223) vs the remaining controls who were noncarriers for V617F (n = 252 140).
For these MPN cases plus V617F carriers, we replicated the germ line JAK2 46/1 haplotype (rs59384377: odds ratio OR = 2.4, P = 6.6 × 10−89), previously associated with V617F-positive MPN. We also identified genome-wide significant associations in the TERT gene (rs7705526: OR = 1.8, P = 1.1 × 10−32), in SH2B3 (rs7310615: OR = 1.4, P = 3.1 × 10−14), and upstream of TET2 (rs1548483: OR = 2.0, P = 2.0 × 10−9).
These associations were confirmed in a separate replication cohort of 446 V617F carriers vs 169 021 noncarriers. In a joint analysis of the combined GWAS and replication results, we identified additional genome-wide significant predisposition alleles associated with CHEK2, ATM, PINT, and GFI1B. All SNP ORs were similar for MPN patients and controls who were V617F carriers. These data indicate that the same germ line variants endow individuals with a predisposition not only to MPN, but also to JAK2 V617F clonal hematopoiesis, a more common phenomenon that may foreshadow the development of an overt neoplasm.
Introduction
Although the JAK2 V617F (V617F) mutation is identified in almost all patients with polycythemia vera (PV) and ∼50% to 60% of patients with essential thrombocythemia (ET) and primary myelofibrosis (PMF),1-4 these BCR-ABL1–negative myeloproliferative neoplasms (MPNs) exhibit a spectrum of phenotypes that cannot be explained by a single molecular abnormality. V617F gene dosage also contributes to phenotype; V617F allele burden tends to be higher in PV and PMF because of acquired uniparental disomy (UPD), whereas a lower allele burden is generally observed in ET patients.1-5
Mutations in exon 12 of JAK2 in PV,6 in the thrombopoietin (TPO) receptor MPL in ET and PMF,7,8 in CBL,9 or in SH2B3 (LNK),10 also result in JAK-STAT pathway activation in MPNs. Somatic deletion and insertion mutations in the calreticulin (CALR) gene were identified in the majority of ET and PMF patients with nonmutated JAK2 or MPL.11,12 MPN heterogeneity is also generated by the acquisition of molecular abnormalities in addition to V617F (eg, TET2, ASXL1, SRSF2, IDH 1/2, DNMT3A, EZH2, and TP53),13 although no strict temporal order of these mutations defines the chronic or blast phase of MPNs.
Individual genetic background may influence the propensity for developing an MPN. A five- to sevenfold increased risk of MPN development was identified in first-degree relatives of patients with MPN.14 Three studies demonstrated that a germ line haplotype (GGCC, referred to as “46/1”) encompassing the 3′ region of the JAK2 gene is associated with a three- to fourfold risk of developing a V617F-positive MPN.15-17 Patients who were heterozygous for this haplotype preferentially acquired the V617F mutation in cis with the predisposition allele (∼75% of cases),15,16 suggesting that the haplotype may lead to hypermutability at the JAK2 locus. However, the haplotype was also weakly associated with V617F-negative or MPL-mutant MPN,15,18-20 suggesting that it may also confer a more generalized propensity for MPN development independent of V617F. Although the 46/1 haplotype contributes to ∼40% to 50% of the attributable risk for MPN development at a population level,16 additional inherited predisposition alleles likely contribute to this increased susceptibility.
In general population studies, the V617F mutation has also been identified in subjects without a diagnosed MPN or other overt hematologic disorder.21-29 Although the clinical significance of V617F positivity (eg, JAK2 V617F-mutant clonal hematopoiesis) in this context is unclear, some of these analyses have begun to characterize time-dependent rates of MPN development as well as the prevalence of non-MPN phenotypes in these individuals.
In this study, we performed genome-wide association analyses in order to identify additional germ line risk factors associated with MPNs, as well as JAK2 V617F clonal hematopoiesis in the general population. This MPN research initiative employed a web-based strategy to recruit a cohort of MPN patients who submitted saliva samples for genotyping, as well as additional customers of the 23andMe Personal Genome Service (PGS) who served as population controls.
Methods
Subjects
Individuals with MPNs (including PV, ET, PMF, post-PV/ET myelofibrosis MF, systemic mastocytosis, chronic myelogenous leukemia, chronic eosinophilic leukemia, and hypereosinophilic syndromes) were invited to participate in a free online MPN research initiative sponsored by 23andMe, Inc (Mountain View, CA) and were additionally provided with the 23andMe PGS at no cost. The 23andMe PGS is a direct-to-consumer genetic testing product of 23andMe, Inc (a more detailed description is provided in supplemental Appendix A, available on the Blood Web site). Customers submit a saliva DNA sample, are genotyped using a genome-wide single-nucleotide polymorphism (SNP) array, and receive a combination of ancestry analyses and reports of genetic contributions to health and other traits.
Participants provided informed consent and participated in the research online under a protocol approved by the external Association for the Accreditation of Human Research Protection Programs–accredited institutional review board, Ethical & Independent Review Services. Between August 2011 and December 2013, we enrolled a total of 1451 participants into this research initiative. We used data from additional customers of the 23andMe PGS who had agreed to participate in research between January 2011 and November 2013 as population controls for genome-wide association study (GWAS). For replication, we used customer data collected between January 2014 and February 2015.
Genotype data
DNA extraction and genotyping were performed on saliva samples by National Genetics Institute, a Clinical Laboratory Improvement Amendments–certified clinical laboratory and subsidiary of Laboratory Corporation of America. Individuals included in the GWAS were genotyped using a custom version of the Illumina HumanOmniExpress+ BeadChip. This platform has a base set of 730 000 SNPs, augmented with a custom set of ∼250 000 SNPs selected by 23andMe. The custom content included 4 probes for the V617F somatic mutation. Samples that failed to reach a 98.5% call rate for SNPs on the standard portion of the array were reanalyzed. Individuals whose analyses failed repeatedly were recontacted by 23andMe customer service to provide replacement samples.
Participant genotype data were imputed against the August 2010 release of 1000 Genomes phase 1 reference haplotypes (refer to supplemental Appendix A for additional details regarding methods; see also http://faculty.washington.edu/browning/beagle/beagle.html, http://genome.sph.umich.edu/wiki/Minimac, and http://www.1000genomes.org/).30-32 Replication samples were genotyped on a fully custom SNP array including 560 000 SNPs selected by 23andMe, with 8 probes for the V617F mutation. This data were imputed against the September 2013 release of 1000 Genomes phase 1 haplotypes, but methods and quality control were otherwise as described for the discovery GWAS.
Phenotype data
Phenotype data were collected by participant self-report using web-based surveys (supplemental Appendix A). Participants in the MPN research initiative filled out an online background survey that included questions about their specific diagnosis, treatment, and current disease status.
Somatic mutation analysis
To estimate V617F mutation burden from SNP array data, we computed a ratio of normalized intensity for the mutant allele to the sum of mutant and wild-type intensities, separately for each of the available probes. Individual observations with unusually low total intensity were removed, and then the median of the remaining observations was used as the mutation burden score.
We validated a subset of results using competitive allele-specific real-time polymerase chain reaction (RT-PCR) (CastPCR, assay JAK2_12600_mu; Life Technologies, Carlsbad, CA). Using serial dilutions of homozygous mutant DNA derived from the UKE-1 cell line (GM23245; Coriell Institute for Medical Research, Camden, NJ) into wild-type human DNA, we verified that the CastPCR assay was sensitive to the mutation at the 0.1% level in a wild-type background. Because of the potential variability of blood cells in saliva, which was used as the source of genotyping instead of peripheral blood or bone marrow, we evaluated the correlation between the allele fraction of JAK2 V617F in saliva and blood in a cohort of MPN patients (supplemental Appendix B).
Statistical analysis
Individuals included in the genome-wide association analysis were selected for having >97% European ancestry, as determined through an analysis of local ancestry by comparison with the 3 HapMap 2 populations.33 A maximal set of unrelated individuals was selected using a segmental identity-by-descent estimation algorithm.34 Individuals were defined as close relatives if they shared >700 cM identity-by-descent, corresponding approximately to the minimal expected sharing between first cousins. Principal components analysis was used to model population substructure.
Genome-wide association tests were performed using imputed allele dosages. Each SNP was tested for association by logistic regression, assuming additive allelic effects. Age, sex, and 5 principal components35 were included as covariates, and P values were computed using a likelihood ratio test. Results were adjusted for residual inflation using genomic control.36
In regions with genome-wide significant associations, we performed stepwise conditional association tests to identify secondary signals. We added terms to the model for the most strongly associated SNP in a region and recomputed likelihood ratio test statistics for all other SNPs in an interval spanning 200 kb beyond any SNP in the region with P < 1 × 10−6. Results were genomic control–adjusted using the inflation factor from the original GWAS. We considered a secondary association to be significant if it had a Bonferroni-adjusted P <.05 accounting for the number of additional tests performed across the entire interval. Supplemental Appendix C lists the study populations that comprise the GWAS analyses included in this report, the genomic control inflation factor for each specified GWAS, and study design considerations. Supplemental Figure 1 displays the Q-Q plots for each GWAS.
Results
Detection of the somatic JAK2 V617F mutation
We investigated whether we could detect the presence of V617F by comparing evidence for the mutation determined from our SNP arrays with self-reported V617F status. The array-based V617F mutation burden strongly correlated with self-reported mutation status (Figure 1A). In population controls, we observed individuals who also had elevated V617F allele ratios, and both the prevalence and distribution of elevated allele ratios were strongly age dependent (Figure 1B). In both groups, higher allele burdens were accompanied by UPD (supplemental Appendix D).
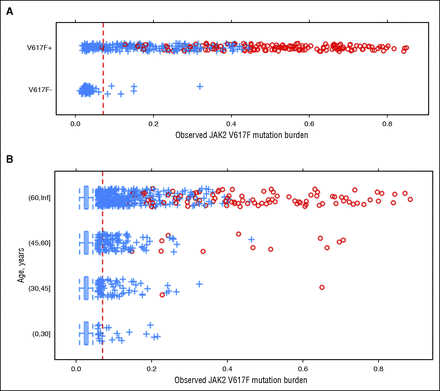
Figure 1
Distribution of JAK2 V617F array-based allele burden scores. Scores should be monotonically related to the proportion of the mutant allele in the DNA sample but were not calibrated. Individuals with evidence of UPD are shown as red circles. (A) Scores among MPN research initiative participants, stratified by self-reported mutation status (V617F-negative – or V617F-positive +). (B) Scores among population controls not enrolled in the MPN research initiative, stratified by age. The red dashed line represents the threshold used to classify V617F carriers in the analysis. Inf, infinity.
We characterized classification performance of the array-based assay for V617F by using self-reported V617F-positive individuals to set a lower bound on the test sensitivity, assuming that any negative result in this group was a false negative. We used younger age population controls (age <40) to set a lower bound on the specificity of the test, on the assumption that any positive result for a younger age control was a false positive. We selected the detection threshold at which we expected a sensitivity of >85% and a specificity of >99.97%, giving an expected positive predictive value of >82% based on the observed distribution of positive results across all ages. Using this prespecified detection threshold, we selected 129 samples for validation using a sensitive RT-PCR assay. Of these, 126 samples tested positive for the V617F mutation, giving a specificity of 99.998% and positive predictive value of 97.7% (see supplemental Appendix E for additional analysis of assay performance). In a separate cohort of 26 MPN patients, we found a strong positive correlation between the allele fraction of V617F in paired samples of saliva and blood (Pearson correlation coefficient, R = 0.95; supplemental Appendix B), indicating the feasibility of using saliva for genotyping of V617F status.
Cohort characteristics
Among the 1451 participants who enrolled in the MPN research initiative, we identified a cohort of 726 unrelated individuals (Table 1) with a self-reported diagnosis of ET (N = 246), PV (N = 258), PMF (N = 87), or related/overlapping MPN diagnoses (designated M-R) including ET/PV, PV + MF, or ET + MF (total N = 135) (supplemental Table 1). This M-R group primarily represented patients with dynamically evolving or overlapping diagnoses of ET and PV, and PV or ET that transformed to MF. Among the 252 637 unrelated population controls, we identified 497 individuals (0.2%) who had evidence for the V617F mutation (V617F carriers). Prevalence of V617F varied from 0.03% under the age of 30, to 0.46% over the age of 60. This group tended to be older than participants in the MPN research initiative (70% over the age of 60 vs 31% of MPN participants). Although MPN initiative participants were predominantly female (69.3%), individuals in the population controls identified by their somatic mutation status were predominantly male (59.4%).
Table 1
GWAS cohort demographics
We tested 342 other self-reported phenotypes for association with V617F carrier status in the population controls, adjusting for age, sex, and genetic principal components (supplemental Tables 2 and 3). A few of these control individuals, although not enrolled in the MPN research initiative, independently reported having an MPN, and this was strongly associated with V617F carrier status (odds ratio OR = 238, P = 5.6 × 10−27). Nonspecific reports of blood disorders were also more common in V617F carriers (OR = 9.78, P = 9.5 × 10−21). V617F carriers were less likely to report having high cholesterol (OR = 0.53, P = 1.4 × 10−8) and had lower body mass index (β = −1.25 kg/m2, P = 5.3 × 10−6). Among other hematologic traits, V617F carriers reported more blood clots (OR = 1.8, P =.014), less anemia (OR = 0.45, P =.019), and higher prevalence of stroke (OR = 1.9, P =.03), but these results were not significant after adjusting for multiple tests. An analysis of reasons V617F+ controls gave for enrolling in 23andMe is provided in supplemental Appendix F.
Genome-wide association analysis
We performed separate genome wide association analyses comparing the 726 MPN cases with all 252 637 population controls (“MPN GWAS”; supplemental Appendix C) and compared the 497 V617F carriers from the control population with the remaining 252 140 V617F negative controls (“V617F status GWAS”; supplemental Appendix C). In both analyses, we observed strong associations with variants in the 9p24.1 region around JAK2 and the 5p22 region in TERT (supplemental Figure 2; supplemental Table 4). An association in the SH2B3 gene was suggestive (P < 1 × 10−6) in the MPN analysis and was genome-wide significant in the analysis of V617F carrier status among population controls.
Motivated by the consistency of results from these 2 analyses, we performed a “combined GWAS” analysis of the 1223 MPN cases plus V617F carriers vs 252 140 V617F noncarriers (Figure 2; supplemental Appendix C). In the combined analysis, 4 loci were genome-wide significant at the P < 5 × 10−8 level (Table 2; supplemental Figure 3). The strongest signal, in the 9p24.1 region, was in the JAK2 gene (rs5938477: OR = 2.46, P = 6.6 × 10−89), consistent with prior reports of association at this locus. Strength of association of the JAK2 variant rs59384377 varied with MPN diagnoses and generally followed the pattern of V617F mutation prevalence across diagnoses, with the strongest effect and highest prevalence in PV and the smallest effect and lowest prevalence in ET (Figure 3; supplemental Figure 4). We observed strong evidence for association extending beyond the end of the JAK2 gene; however, the SNPs with the strongest effects were near the 5′ end of the gene, in the second intron. After adjusting for the effect of rs59384377 (supplemental Figure 5; supplemental Table 5), we observed a secondary association in the same region (rs10974900, P = 6.2 × 10−6), 17 kb from rs59384377.
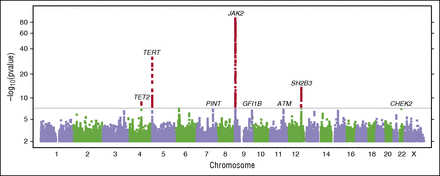
Figure 2
Manhattan plot of the combined GWAS of MPN cases and V617F carriers. Results with P < 5 × 10−8 (conventional threshold for genome-wide significance) are shown in red. We have also labeled suggestive results (P < 1 × 10−6) discussed in the text. Gene labels are provided for cross referencing with other results and are not intended to suggest a causal basis for the observed associations.
Table 2
Association statistics for lead SNPs
SNP associations with specific MPN diagnoses vs population controls. Here, the “case” result represents all cases used in the GWAS.
The second significant locus was in TERT, the gene encoding the protein component of telomerase reverse transcriptase. TERT rs7705526 was associated with an OR of 1.8 per G allele, P = 2.9 × 10−32. TERT variant rs7705526 was associated with ET, PV, and MF with similar effect sizes. We identified a secondary association at TERT rs2853677 (supplemental Figure 6; supplemental Table 5) that remained genome-wide significant after conditioning on rs7705526 (P = 1.1 × 10−12). We identified a third association with lead SNP rs7310615 in the SH2B3 gene (OR = 1.5, P = 3.1 × 10−14) in strong linkage disequilibrium (r2 = 0.94) with nonsynonymous variant rs3184504, W262R. The fourth identified association was with the lead SNP rs1548483 (OR = 2.0, P = 2.0 × 10−9), which falls 300 kb upstream of the TET2 gene. In a secondary analysis of other MPN diagnoses not included in the GWAS (supplemental Table 6), rs1548483 was nominally associated with chronic myelogenous leukemia (N = 130, P =.034, OR = 2.1) and systemic mastocytosis (N = 210, P =.013, OR = 2.0), other diagnoses enlisted within the MPN research initiative.
We observed suggestive association signals with relatively large effect sizes at low-frequency nonsynonymous variants in CHEK2 (1100delC, rs555607708: OR = 4.4, P = 7.5 × 10−8) and ATM (F858L, rs1800056: OR = 2.2, P = 6.5 × 10−7), with consistent effects in both MPN cases and V617F carriers. Two additional suggestive associations with smaller effect sizes from this combined GWAS included rs58270997 in the long intergenic noncoding RNA PINT (P = 1.1 × 10−7), and rs621940 downstream of the gene for growth factor independent 1B (GFI1B) (P = 1.9 × 10−7) (Table 2). We tested all pairs of index SNPs for interactions, but none were significant after adjusting for multiple testing (supplemental Appendix G).
We tested whether each index SNP could discriminate between V617F-positive or V617F-negative MPNs, using a case-only approach. Although JAK2 rs1327494 was more strongly associated with V617F-positive disease (OR = 2.7, P = 4.5 × 10−18), TERT rs7705526 was only nominally associated with V617F-positive disease (OR = 1.3, P=.04). TERT rs2853677, SH2B3 rs7310615, and TET2 rs1548483 were not significantly associated with V617F status in MPN cases (Table 3; all P >.05). However, we observed a trend toward larger effect sizes for V617F-positive cases than for V617F-negative cases vs controls (supplemental Figure 7).
Table 3
Association of SNPs with JAK2 V617F positivity in MPN cases
Replication analysis
Using the same array-based detection method, we identified 446 JAK2 V617F carriers in an additional set of 169 467 unrelated individuals (supplemental Table 7; supplemental Appendix C). In this set, we confirmed associations between V617F status and lower risk of high cholesterol (OR = 0.59, P= 3.9 × 10−5), lower body mass index (β = −1.4 kg/m2, P = 4.5 × 10−6), and higher risk of blood clots (OR = 2.3, P = 9.3 × 10−6). This replication analysis of 446 JAK2 V617F carriers vs 169 021 noncarriers confirmed associations with similar effect sizes that were observed from the combined GWAS analysis (Table 4).
Table 4
Replication cohort results for lead SNPs
We combined results from the combined GWAS and replication cohorts, using Fisher’s method to compute joint P values. The 4 suggestive associations described for the combined GWAS analysis (ATM, CHEK2, PINT, and downstream of GFI1B) reached genome-wide significance in this joint analysis (Table 4, Joint P column). In the combined cohorts, we tested for associations between clinical phenotypes and either V617F allele burden or UPD status, and found that the associations seen for V617F presence tended to be stronger at higher allele burdens or with UPD. Among our GWAS loci, the risk allele for TERT rs59384377 was associated with lower allele burden and risk of UPD, and the risk allele for GFI1B rs621940 was associated with higher burdens and risk of UPD (supplemental Appendix H).
Discussion
Our study extends the understanding of the genetic predispositions underlying BCR-ABL1–negative MPNs and JAK2 V617F clonal hematopoiesis in the general population. In addition to the 46/1 JAK2 haplotype, we identify predisposition alleles associated with TERT, SH2B3, TET2, ATM, CHEK2, PINT, and GFI1B in both MPN patients and in population controls who are V617F carriers. These genes impact diverse biologic pathways such as cellular aging (TERT), JAK-STAT signaling (JAK2, SH2B3), epigenetic regulation (TET2), DNA damage repair and/or tumor suppressor function (ATM, CHEK2, PINT), and erythroid/ megakaryocyte development (GFI1B).
Age-related clonal hematopoiesis in the general population was recently found to be associated with adverse outcomes, including an increased risk of hematologic cancer and all-cause mortality.27-29,37JAK2 was among the most commonly mutated genes in these studies, which also included DNMT3A, TET2, ASXL1, and TP53. The predisposition alleles we have identified in our study have similar ORs for V617F-positive and V617F-negative MPNs, as well as JAK2 V617F clonal hematopoiesis in unselected population controls.
Besides the replicated JAK2 46/1 haplotype, the second most statistically significant SNPs in our analysis were 2 TERT loci, rs7705526 and rs2853677, previously identified in several cancer GWAS studies.38-41 Since our initial identification of TERT (eg, SNP rs2853677) as a predisposition allele for MPNs,42 another germ line variant in TERT, rs2736100, was identified in Icelanders diagnosed with MPN.43 Our lead SNP, rs2853677, is in moderate linkage disequilibrium with rs2736100 (r2 = 0.54), and we see similar evidence for association at rs2736100 (OR = 1.6, P = 7.5 × 10−13). SNP rs2736100 has previously been associated with increased red blood cell and platelet counts and white blood cells of the myeloid lineage.43 Recently, a third study corroborated TERT as a predisposition gene for MPN.44
Our association with rs7310615 in the SH2B3 gene, in strong linkage disequilibrium with nonsynonymous variant rs3184504, or SH2B3 W262R, corroborates prior data showing an association between W262R and MPN patients.45 Relevant to MPNs, SH2B3 binds to the TPO receptor MPL; upon cytokine stimulation with TPO, SH2B3 also binds strongly to JAK2 and inhibits downstream STAT activation, resulting in negative feedback.46,47SH2B3 −/− mice exhibit an MPN phenotype, including an expanded hematopoietic stem cell compartment with increased self-renewal, megakaryocytic hyperplasia, splenomegaly, leukocytosis, and thrombocytosis.48 Somatic mutations in SH2B3 have been identified at low frequency in MPN10,12 and result in aberrant JAK-STAT activation.10
This study’s identification of a predisposition allele associated with TET2 is intriguing given the common finding of somatic TET2 mutations in a spectrum of myeloid malignancies, including MPN.49,50 In MPN, TET2 mutations can be identified in hematopoietic stem cells and can either precede or follow the acquisition of V617F.13,49 The mutational order of these 2 genes can influence the clinical and biologic behavior of these neoplasms.51 Mutations in TET2 constitute one of the early molecular lesions in myeloid neoplasms, including MPN; similarly, it is one of the more commonly mutated genes in individuals with clonal hematopoiesis.27-29,52
In the joint analysis results of MPN cases plus V617F carriers vs noncarriers of V617F from the 2 unrelated control populations, genome-wide significance was observed for the variants CHEK2 1100delC and ATM F858L, as well as PINT and GFI1B. ATM and the G2 checkpoint kinase CHEK2 are closely related components of the DNA-damage response pathway, which plays a critical role in maintaining genomic integrity. Mutations in genes of this pathway compromise DNA repair and are linked to several types of malignancies and heritable cancer syndromes,53-56 Although the germ line variants CHEK2 1100delC and ATM F858L have been associated with chronic lymphocytic leukemia,57 sparse data exist for their relationship to myeloid malignancies, including MPN.58,59 Somatic mutations in CHEK2 were recently identified in 3 of 151 MPN patients,12 highlighting a potential role of this gene in MPN pathogenesis. The long intergenic noncoding RNA PINT is regulated by p53 and interacts with polycomb repressive complex 2, a key regulator of hematopoietic stem cell differentiation and maintenance frequently inactivated in myeloid malignancies.60 GFI1B is a zinc-finger transcriptional repressor that is essential for erythropoiesis and megakaryopoiesis.61 A nonsense mutation in GFI1B was identified in a family with autosomal dominant gray platelet syndrome.62 The truncated GFI1B mutant protein acts in a dominant-negative manner to block the normal development of megakaryocytes and platelets.
A recent study has reported additional associations with MPN at 3q26.2 downstream of MECOM, and at 6q23.3 between HBS1L and MYB.44 At 3q26.2, we find weak support for the reported association at rs2201862 (GWAS P =.02, OR = 0.91 for T allele) but more evidence at an intronic site in MECOM (rs3851379: GWAS P = 2.7 × 10−7, OR = 1.24 for G allele; replication P =.044, OR = 1.15; joint P = 2.3 × 10−7). At the 6q23.3 locus, we replicate the reported association (GWAS P =.0040, OR = 1.14 for A allele; replication P =.27, OR = 1.09; joint P =.0085).
Studies have reported the detection of chromosomal abnormalities in blood, saliva, or buccal samples from SNP array data.63,64 However, these investigations did not report detection of somatic point mutations, largely because these variants are not present on standard genome-wide genotyping arrays designed to assay germ line variation. We show that it is feasible to detect the V617F mutation from SNP array data with high sensitivity and specificity. Although we do not expect this test to match the sensitivity of targeted blood-based assays, it may remain attractive as a screen followed by secondary testing. Although the variable composition of DNA in saliva from blood may affect the sensitivity of this assay, we demonstrated a very strong correlation between the V617F allele fraction in the peripheral blood and saliva of individual MPN patients. In addition to its application to blood and bone marrow, RT-PCR quantification of V617F mutant allele burden in saliva merits further exploration.
The 0.2% rate of V617F positivity in our control population mirrors the rates of 0.14% and 0.17% found in 2 Copenhagen general population studies,22,26 and the rates of 0.19%27 and 0.18%28 in 2 studies of age-related clonal hematopoiesis. In our study, the median age of V617F carriers was 68 years, with the incidence rising from 0.03% for ages 21 to 30 to 1.1% for ages 81 to 90. In our analysis and the Copenhagen General Population Study,22 V617F carrier status was more frequently associated with male sex, increasing age, and a strikingly similar multifactorial-adjusted OR for MPN (OR = 221 for the Copenhagen study; OR = 238 for the current GWAS). In addition, ORs for venous thromboembolism and deep venous thrombosis were 3.1 and 4.6 in the Copenhagen study; in our analysis, V617F carriers were similarly more likely to report a history of blood clots (OR = 1.8, P =.014). The 0.2% prevalence rate (age-adjusted: 0.16%) of JAK2 V617F in the control cohort is roughly double the estimated age-adjusted prevalence rate of combined JAK2 V617F-positive MPN (PV+ ET + PMF + post-PV MF+ post-ET MF) in the United States based on data from 2 large health care plans.65 Therefore, the excess prevalence of JAK2 V617F clonal hematopoiesis among unselected controls likely represents a combination of (1) persons with an undiagnosed MPN, (2) individuals who may develop a future MPN, and (3) and those who will never develop a hematologic disorder.
In summary, we have identified inherited loci in or near TERT, SH2B3, TET2, ATM, CHEK2, PINT, and GFI1B that predispose to both age-related JAK2 V617F clonal hematopoiesis in the general population, as well as MPN independent of V617F status. In particular, JAK2, SH2B3, TET2, and CHEK2 represent 4 genes wherein both inherited variation and somatic mutation have been found to contribute to V617F clonal hematopoiesis and/or MPN development. The use of SNP arrays with custom probes for specific somatic mutations can be applied to analyses of other genes implicated in age-related clonal hematopoiesis and hematologic cancers.


The 23andMe PGS is a direct-to-consumer genetic testing product of 23andMe, Inc (a more detailed description is provided in supplemental Appendix A, available on the Blood Web site). Customers submit a saliva DNA sample, are genotyped using a genome-wide single-nucleotide polymorphism (SNP) array, and receive a combination of ancestry analyses and reports of genetic contributions to health and other traits.
In my opinion you commit an error. I can defend the position. Write to me in PM, we will talk.