
Idelalisib is a first-in-class oral inhibitor of PI3Kδ that has shown substantial activity in patients with relapsed/refractory chronic lymphocytic leukemia (CLL). To evaluate idelalisib as initial therapy, 64 treatment-naïve older patients with CLL or small lymphocytic leukemia (median age, 71 years; range, 65-90) were treated with rituximab 375 mg/m2 weekly ×8 and idelalisib 150 mg twice daily continuously for 48 weeks.
Patients completing 48 weeks without progression could continue to receive idelalisib on an extension study. The median time on treatment was 22.4 months (range, 0.8-45.8+). The overall response rate (ORR) was 97%, including 19% complete responses.
The ORR was 100% in patients with del(17p)/TP53 mutations and 97% in those with unmutated IGHV. Progression-free survival was 83% at 36 months. The most frequent (>30%) adverse events (any grade) were diarrhea (including colitis) (64%), rash (58%), pyrexia (42%), nausea (38%), chills (36%), cough (33%), and fatigue (31%). Elevated alanine transaminase/aspartate transaminase was seen in 67% of patients (23% grade ≥3). The combination of idelalisib and rituximab was highly active, resulting in durable disease control in treatment-naïve older patients with CLL. These results support the further development of idelalisib as initial treatment of CLL. This study is registered at ClinicalTrials.gov as #NCT01203930.
Introduction
Chronic lymphocytic leukemia (CLL) has a B-cell immunophenotype and accounts for one-third of all leukemias.1 In the United States, it is estimated that ∼15 720 people were diagnosed with CLL and 4600 died of the disease in 2014.2 It mainly affects older adults, with an average age of 72 years at the time of diagnosis.3 Symptoms may include fever, night sweats, or weight loss, often accompanied by lymphadenopathy, splenomegaly, or hepatomegaly, as well as cytopenias.4,5
Chemoimmunotherapy is the standard initial treatment of patients with CLL.6 However, this therapy is associated with substantial toxicities, including myelosuppression and infections, which can be especially problematic in older patients with significant infirmities or baseline cytopenias.7,8 A variety of alternate therapies are available for these patients, but they are generally less effective than chemoimmunotherapy.9 Treatments with high efficacy and less toxicity are needed for this population.
The B-cell receptor (BCR) pathway is important in both normal and malignant B cells.10 Signaling through the BCR provides a strong proliferative and survival stimulus to the cell; interfering with such signaling is therefore a rational approach to the treatment of B-cell malignancies. BCR signaling is mediated partly by the activation of phosphoinositide 3-kinase (PI3K).11 The δ isoform, containing the p110δ catalytic subunit, is one of the 4 PI3K class I enzymes and is the most important isoform involved in CLL.12-14 Activation of PI3Kδ subsequently activates the serine/threonine kinases AKT and mammalian target of rapamycin (mTOR) and exerts multiple effects on cell migration, metabolism, proliferation, differentiation, and survival.15,16
Idelalisib is a potent, highly selective, oral small-molecule inhibitor of PI3Kδ.13,17,18 In phase 1 studies, idelalisib alone and in combination with other therapies demonstrated significant clinical efficacy with an acceptable toxicity profile in patients with relapsed and refractory CLL.19,20 Idelalisib, in combination with the anti-CD20 antibody rituximab, was approved by the US Food and Drug Administration in July 2014 for the treatment of relapsed CLL based on a pivotal phase 3 trial.21 Rituximab monotherapy has also shown activity when administered to patients with previously untreated CLL.22,23
Given the role of PI3Kδ in B-cell malignancies, the pharmacologic profile of idelalisib, the minimal myelosuppression and pronounced efficacy seen with idelalisib in patients with relapsed/refractory CLL, and the activity of rituximab in treatment-naïve patients, we performed a phase 2 open-label study of idelalisib in combination with rituximab in older patients with previously untreated CLL.
Patients and methods
The present study was initiated in September 2010 (Study 101-08; ClinicalTrials.gov #NCT01203930). Of the 64 total patients in the trial, 59 with CLL and 5 with small lymphocytic leukemia (SLL) were enrolled. The primary trial evaluated patients through 48 weeks of idelalisib treatment followed by an extension study offering continued therapy for patients who were deriving clinical benefit (Study 101-99; ClinicalTrials.gov #NCT01090414).
The primary study was completed in March 2013 and the extension study is ongoing. This analysis is based on data up to August 2014. Both trials were conducted according to principles of Good Clinical Practice at 5 centers in the United States after review and approval by the US Food and Drug Administration. Institutional review boards at each study site approved the protocols. All patients provided written informed consent. All authors had full access to study data and were involved in data interpretation, manuscript preparation, revision, and final approval.
The primary objective of this study was to evaluate the overall response rate (ORR) of idelalisib when combined with rituximab in older patients with previously untreated CLL or SLL. The secondary objectives were to assess the duration of response (DOR), progression-free survival (PFS), safety, pharmacokinetics, and pharmacodynamics of idelalisib plus rituximab in this patient population.
Eligibility criteria
Older patients with histologically or cytologically confirmed CLL or SLL as established by the International Workshop on CLL (IWCLL)24 were eligible for the study. Patients were enrolled if they met all of the following criteria: (1) age ≥65 at the first visit; (2) Binet stage C or Rai stage III or IV CLL or active disease, or active SLL, defined per IWCLL 2008 criteria; and (3) World Health Organization performance status of ≤2. Disease-related cytopenias of any grade were permitted at study entry.
Patients were excluded from the study for any of the following reasons: (1) prior therapy for CLL or SLL, except corticosteroids for symptom relief; (2) known active central nervous system involvement with malignancy; (3) ongoing active, serious infection requiring systemic therapy; (4) serum creatinine ≥2.0 mg/dL; (5) serum bilirubin ≥2.0 mg/dL (unless caused by Gilbert syndrome) or serum transaminases (ie, aspartate aminotransferase AST, alanine aminotransferase ALT) ≥2× upper limit of normal, (6) positive test for human immunodeficiency virus antibodies; or (7) active hepatitis B or C (patients with serologic evidence of prior exposure but with negative DNA or RNA testing were eligible). Concomitant treatments for adverse events (AEs), intercurrent illness, or symptom management deemed medically necessary by the investigator were allowed. Medications that were clinically relevant inhibitors or inducers of cytochrome P450 (CYP3A4) were discouraged because preliminary data indicated that they might affect idelalisib plasma exposures.
Study treatments
All patients received idelalisib 150 mg twice daily orally as capsules or tablets on days 1 through 28 of each 28-day cycle for 48 weeks and rituximab 375 mg/m2 IV weekly for 8 doses (cycles 1 and 2). The idelalisib doses were based on safety, efficacy, and pharmacokinetic data from phase 1 studies in healthy volunteers25 and patients with hematologic malignancies.20 The rituximab regimen was previously shown to be safe in a population of patients with relapsed and refractory SLL and follicular lymphoma.26 Unless disease progression or intolerable toxicity occurred, patients continued idelalisib through 48 weeks on the primary study and indefinitely on the extension study.
Idelalisib doses were taken at ∼12-hour intervals with water, with or without food. Rituximab was administered IV according to the study site’s usual practice and in accordance with the prescribing information. Premedication with an analgesic/antipyretic and an antihistamine was administered before each infusion. Premedication with glucocorticosteroids for the first dose, and as indicated for subsequent infusions, was allowed.
Study assessments
Clinical efficacy was evaluated with physical examination or computed tomography (CT) scans after study weeks 8, 16, 24, 36, and 48 according to standard criteria.24 The ORR was assessed by each investigator and was defined as the proportion of patients who achieved a complete response (CR) or partial response (PR). Lymph node response rate was assessed and defined as the proportion of patients with a ≥50% decrease in the sum of the products of the greatest perpendicular diameters (SPD) of index lesions. DOR (the time from the date of initial response to the date of documented disease progression or death, whichever occurred first) and PFS (the time from the date of the first dose of study treatment in cycle 1 to the date of death or classification of progressive disease, whichever occurred first) were evaluated.
The change from baseline in the SPD of index lesions and response rates for splenomegaly, hepatomegaly, absolute lymphocyte count (ALC), platelets, hemoglobin, and absolute neutrophil count (ANC) were also measured. Safety was assessed by monitoring of physical examination, vital signs, clinical laboratory tests (hematology, serum chemistry, coagulation, and urinalysis), electrocardiogram, and AEs. Toxicity of AEs was graded according to the Common Terminology Criteria for Adverse Events (CTCAE, versions 4.02 and 4.03).
Cancer Genetics, Inc. (Rutherford, NJ) analyzed extracted genomic DNA from frozen baseline blood samples to assess CLL-related genetic abnormalities.27 The presence of 17p deletions and/or TP53 mutations were examined in relation to the efficacy end points (DOR, PFS, time to response TTR). For analysis of the immunoglobulin heavy-chain variable region (IGHV), sequences within the IGHV leader sequence and IGHJ region were considered unmutated if homology to the corresponding germline gene was ≥98%. For TP53 mutation analysis, exons 5-6 and 7-9 were amplified and mutations were documented if present by bidirectional sequencing. Loss of 17p was determined by comparative genomic hybridization using a custom oligonucleotide array; samples were considered positive for del(17p) when >50% of chr17:6.0-9.0 Mbp was deleted and if confirmed by quantitative polymerase chain reaction (ratio of ≤0.8 for TP53 relative to control genes TERT, RAG2).
Patients who developed progressive disease at any point stopped study drug treatment and discontinued the study. Those deriving clinical benefit were given the option to enroll in the long-term safety extension protocol in which treatment with idelalisib was continued.
Statistics
Efficacy and safety end points were analyzed for patients who received at least one dose of study drug. Response rates were computed with exact 95% confidence intervals (CIs). DOR and PFS were estimated using the Kaplan-Meier (KM) methods, censoring for patients without progression events at the date of last tumor assessment. Patient incidence of treatment-emergent adverse events (TEAEs) and laboratory abnormalities were summarized with descriptive statistics. Unless otherwise noted, data from the primary study and the extension study were considered together.
Results
Patient characteristics
Beginning in September 2010, 64 patients with previously untreated CLL or SLL were enrolled in the study. Patient demographics and disposition are summarized in Table 1. The majority of patients were representative of the typical CLL population: older, predominantly male, and Caucasian. In addition, 40.6% of patients exhibited common CLL symptoms, including night sweats (26.6%) and fatigue (21.9%). The majority of patients had good performance status: 56.3% had a World Health Organization performance status score of 1 and 42.2% had a score of 0. The median time from diagnosis was 3.3 years (range, 0.1-11.3). Prognostic genetic findings at baseline are shown. Three patients did not consent to DNA sample collection. For subgroup efficacy analyses, only patients with 17p deletions, TP53 mutations, or IGHV mutations were analyzed.
Table 1
Patient baseline characteristics and disposition
Patient disposition
All patients in the intent-to-treat analysis set (N = 64) received idelalisib: 62 patients (96.9%) completed rituximab dosing, and 43 patients (67.2%) completed the primary study (Table 1). Forty-one patients (64.1%) continued treatment in a long-term extension study. The 64 patients received idelalisib for a median of 22.4 months (range, 0.8-45.8) in the combined primary and extension studies. As of August 2014, 23 subjects were still receiving the drug with a median treatment duration of 36.4 months.
Seven significant protocol deviations were reported: 5 of these were a result of continuing the study drug (idelalisib or rituximab) after experiencing grade ≥3 laboratory abnormalities (ALT/AST elevation in 3 patients, elevated creatinine and uric acid in 1, and hypercalcemia in 1, all of which recovered to normal levels). The primary reason for withdrawal from the study as indicated by the investigator was an AE, occurring in 29 patients (19 in the primary and 10 in the extension study).
Safety profile
The incidence of TEAEs occurring in ≥15% of patients is listed in Table 2. All 64 patients in the combined primary/extension studies experienced at least one TEAE and 57 patients (89.1%) had AEs with a severity of grade ≥3, attributed by the investigator to idelalisib in 47 patients (73.4%). The events reported most frequently with a severity of grade ≥3 were diarrhea or colitis (27 patients, 42.2%) and pneumonia (12 patients, 18.8%). There were no cases of Richter’s transformation reported during the study.
Table 2
Incidence of TEAEs (≥15% of patients) and SAEs (≥2 patients), combined primary and extension studies (N = 64)
Twenty-nine patients (45.3%) in the combined primary and extension studies had ≥1 TEAE leading to idelalisib dose reduction to 100 mg twice daily or 75 mg twice daily, most frequently as a result of transaminase elevation (21 patients, 32.8%) and diarrhea or colitis (5 patients, 7.8%). Twenty-nine patients (45.3%) permanently discontinued treatment with idelalisib because of TEAEs. Diarrhea (8 patients, 12.5%), colitis (4 patients, 6.3%), dyspnea (3 patients, 4.7%), and rash (3 patients, 4.7%) were the most frequently reported AEs leading to discontinuation of idelalisib therapy. (See supplemental Table 1 for the full list of AEs leading to idelalisib discontinuation, available on the Blood Web site.)
A total of 42 patients (65.6%) in the combined primary/extension studies experienced at least one serious adverse event (SAE). The most frequently reported SAEs (>5%) were diarrhea or colitis (24 patients, 37.5%) and pneumonia (11 patients, 17.2%). Four patients died during the primary study as a result of AEs: pneumonitis (2 patients), metastatic malignant melanoma (1), and sepsis (1, occurring on day 151 in an 86-year-old man with CLL, regarded as unrelated to idelalisib). An additional patient died after myocardial infarction during the extension study. The 2 cases of pneumonitis were considered by the investigators to be possibly related to idelalisib, and neither coincided with diarrhea/colitis or transaminase elevation. These included 1 patient with SLL, a 72-year-old woman, who was diagnosed with pneumonitis on day 129 of the study and died 17 days later, and a 74-year-old man with CLL who was diagnosed with pneumonitis on day 89 and died 6 days later. In both cases, bronchoscopy was negative for infectious etiology. Two patients developed pulmonary fibrosis; both cases were regarded by the investigator as possibly related to idelalisib. Both events completely resolved (within 2 and 4 weeks, respectively) with the discontinuation of idelalisib and without steroids. Fourteen patients (for whom comedication data are available) experienced pneumonia in the primary study; 8 had been receiving valacyclovir prophylaxis and 1 had been receiving trimethoxazole. The only opportunistic infection was cytomegalovirus in a patient taking valacyclovir. That patient, and one other, received steroids as part of the pneumonia treatment. None of these cases were regarded as drug related.
Eighteen patients reported grade ≥3 diarrhea and 16 had grade ≥3 colitis; 7 patients were reported with both terms. Generally, the term colitis was applied if a patient had a colonoscopic biopsy (performed in at least 14 patients), which in most cases showed inflammation ranging from mild to severe. The median time to grade ≥3 diarrhea or colitis was 9 months (range, 3-29) and appeared in 16 and 11 patients during the primary and extension studies, respectively.
Table 3 summarizes the change from baseline through week 48 in hematologic and clinical chemistry parameters during the primary study. The incidence of serum ALT and/or AST elevation was 67%, with 23% being grade ≥3. The median time to onset for grade ≥3 serum ALT/AST elevation was 1.4 months (range, 0.9-3.0) and the median time to resolution was 1.0 month (range, 0.3-1.3). Serum ALT/AST increases were not associated with any increases in serum bilirubin to > 2× upper limit of normal. Of 15 patients with grade ≥3 transaminase elevation, 3 discontinued idelalisib without an attempt at re-challenge: one after grade 3 ALT/AST alone, one after concurrent grade 3 rash and grade 3 ALT, and one after concurrent grade 3 anemia and grade 3 ALT. The remaining 12 patients were re-challenged after interruption; 11 patients were able to remain on idelalisib at doses of 150 mg twice daily (2 patients), 100 mg twice daily (6 patients), or 75 mg twice daily (3 patients).
Table 3
Incidence of hematologic and nonhematologic laboratory abnormalities (≥15% of patients), primary study
Efficacy
Table 4 summarizes the response rates observed in the study. In the primary study, the ORR for all patients (N = 64) was 96.9%: 9 patients (14.1%) had a CR and 53 patients (82.8%) had a PR. The ORR remained at 96.9% in the combined primary/extension study, but 3 patients with a best response of PR in the primary study achieved a CR in the extension study, bringing the overall CR rate to 18.8% (12 patients; median time to CR was 8.4 months). No patients achieving CR experienced subsequent progression. The ORR was 100% (CR, 33%; PR, 67%) in patients with del(17p)/TP53 mutations and 97% (CR, 8%; PR, 89%) in those with unmutated IGHV.
Table 4
Overall response rate, combined primary and extension studies (N = 64)
Figure 1 shows the best on-treatment percent change in nodal SPD based on serial measurements using either CT scan or physical examination (investigator’s choice). Physical examination was used in 68% (34/50) of evaluable patients, CT scan was used in 10% (5/50), and the method was not recorded in 22% (11/50). All 50 patients with lymph node enlargement at baseline and who had at least one post-baseline efficacy assessment had a reduction in tumor size. Organ-specific responses were achieved in 27 of 28 patients (96%) with splenomegaly and in 7 of 7 patients (100%) with hepatomegaly. In addition, 24 of the 26 patients (92%) who had B symptoms responded by week 16.

Figure 1
Nodal response to treatment with idelalisib and rituximab by patient, in primary study. Best nodal response for 50 patients evaluable for lymph node response (14 inevaluable: 12 from lack of baseline adenopathy and 2 from discontinuation before 8 weeks) (black bars) and patients with either del(17p) or TP53 mutations (N = 5; 4 inevaluable patients not shown) (gray bars). Response assessed by physical examination or CT scan according to standard criteria.24 SPD, sum of the products of the perpendicular diameters of measured lymph nodes.
Figure 2A shows improvement in median hemoglobin and platelet values in those patients with anemia (n = 17) or thrombocytopenia (n = 17) at baseline in the primary study. Normalization of neutrophil counts in those patients with neutropenia (n = 5) at baseline was also observed. Hematologic responses occurred as follows: 53 of 53 patients (100%) for ALC (reduction to <4000/µL or ≥50% from baseline), 16 of 17 patients (94%) for platelet count (increase to >100 000/µL or by ≥50% from baseline), 17 of 17 patients (100%) for hemoglobin (increase to >11.0 g/dL by ≥50% from baseline), and 5 of 5 patients (100%) for ANC (increase to >1500/µL or by ≥50% from baseline).
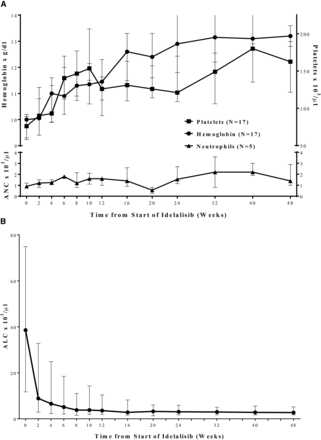
Figure 2
Improvement in median (Q1, Q3) hematologic parameters (A) and lymphocyte counts (B) over time, in primary study. Squares represent platelet count (n = 17); circles represent hemoglobin (n = 17); triangles represent neutrophils (n = 5). ALC, absolute lymphocyte count; ANC, absolute neutrophil count.
Among the 62 responding patients in this study, the median TTR was 1.9 months (range, 1.6-5.7). The median TTR was not affected by del(17p)/TP53 mutation or IGHV mutation status. The KM estimate of median DOR (based on patients achieving a CR or PR) was not reached for this analysis. Figure 3A depicts PFS overall and in patients with del(17p)/TP53 mutations. The KM estimate of median PFS was not reached at the time of this analysis; the PFS at 12, 18, and 24 months was 92.9% and at 36 months, the PFS (95% CI) was 82% (64%, 92%). Four patients (6.3%) had disease progression. As shown in Figure 3B, the median overall survival (OS) has also not yet been reached; the OS (95% CI) at 36 months was 90% (82%, 99%).
PFS (A) and OS (B) in all patients (thick line, N = 64) and those with either del(17p) or TP53 mutation (thin line, N = 9), in combined primary and extension studies. Median PFS and median OS for both groups have not been reached. PFS, progression-free survival; OS, overall survival.
Discussion
Results from this phase 2 trial demonstrate that the combination of idelalisib and rituximab produced a very high ORR (97%) in previously untreated patients with CLL/SLL who were ≥65 years of age. Only 2 of 64 patients did not achieve at least a partial remission with the therapy, both discontinuing as a result of toxicity before the first evaluation time point. Complete remissions were seen in 19% of patients.
Time to response was rapid with idelalisib and rituximab. Unlike prior experience with single-agent idelalisib,20 marked lymphocytosis was not apparent with the combination, likely because of the rapid reduction of lymphocytosis from the addition of rituximab. Median PFS has not been reached thus far, and only 4 patients have relapsed on study.
The current trial was specifically designed as initial therapy for older patients with CLL. The rationale for targeting this population was the lack of tolerability of chemoimmunotherapy in older patients, many of whom have multiple comorbidities.3,28 The primary complications of chemoimmunotherapy are myelosuppression and infection (eg, in a recent trial, 35% and 27% grade ≥3 neutropenia was observed in patients treated with obinutuzumab + chlorambucil and rituximab + chlorambucil, respectively).29 In the current study, although grade ≥3 neutropenia was reported in 28% of patients, nearly all patients with baseline cytopenias showed improvement in cytopenias over time. Thus, the treatment was not significantly myelosuppressive.
Prognostic factors associated with limited responses to chemoimmunotherapy did not affect the response rate of idelalisib and rituximab. Of note, 9 patients were enrolled with either a 17p deletion, a TP53 mutation, or both. All nine of these patients responded to therapy and 6 continued to receive treatment on the extension study. The positive efficacy results with idelalisib therapy in patients with 17p deletion or TP53 mutation are consistent with salutary effects of idelalisib treatment in patients with relapsed/refractory CLL who had these genetic abnormalities.18,20 The proportion of patients with a 17p deletion or TP53 mutation was smaller than that seen in the prior trials of idelalisib in relapsed/refractory CLL but is consistent with the expected frequency of such abnormalities in previously untreated patients.30,31
The most common adverse side effects in the trial were an increase in the transaminases and diarrhea/colitis. The incidence of ALT and/or AST elevation in the current population was 67% with 23% grade ≥3. Diarrhea or colitis was reported in 64% of patients with 42% grade ≥3. These are higher incidences than those observed with single-agent idelalisib in 54 patients with relapsed/refractory CLL/SLL treated in a dose-escalation study,20 in which ALT/AST elevation occurred in 28% (grade ≥3, 2%), and diarrhea/colitis was reported in 30% (grade ≥3, 6%).
A question that arises is whether the increased toxicity is related to the addition of rituximab to idelalisib. Two prior studies using a combination of idelalisib and rituximab provide AE reporting for comparison. In an update to the published randomized trial in relapsed/refractory patients with CLL/SLL (median prior regimens, 3) that compared the combination of idelalisib and rituximab with rituximab and placebo,21,32 the incidence of transaminase elevation was 40% (grade ≥3, 8%) of patients treated with idelalisib and rituximab. The incidence of diarrhea or colitis with the combination was 21% (grade ≥3, 6%). The median duration of idelalisib treatment in this randomized study was, however, only 5 months at the time of analysis, limiting the comparison. In a phase 1b study of 40 patients with relapsed or refractory CLL receiving idelalisib with either rituximab or ofatumumab (median prior regimens, 2), the median duration of idelalisib exposure was 18 months.19 In that trial, the incidence of ALT and/or AST elevation was 30% (grade ≥3, 10%), and diarrhea or colitis occurred in 53% (grade ≥3, 18%).
Based on these data, the increased incidence of toxicity in the current trial may be related to the study population being previously untreated. This is noteworthy because previously untreated patients typically experience significantly less toxicity with chemotherapy-based regimens. To address the relative importance of rituximab vs a treatment-naïve patient population, previously untreated patients ≥65 years old are now being enrolled in an idelalisib monotherapy cohort of the trial. Further analysis of this group will be important in differentiating the potential causes of the increased toxicity observed in this previously untreated cohort.
In this study, infiltration of T lymphocytes was noted in colonoscopic biopsies done for evaluation of diarrhea. Whether this was an epiphenomenon or causal of the underlying symptoms is unclear. T-cell levels are typically normal in previously untreated patients with CLL but are quite low in patients with relapsed/refractory disease, which suggests that the infiltrate could have caused the symptoms. Consistent with this hypothesis, data have emerged showing PI3Kδ to be relevant in the functioning of regulatory T cells,33 which are important in maintaining self-tolerance. In a study of mice with inactivation of P13Kδ caused by a point mutation, the animals developed inflammatory bowel disease limited to the large intestine.34 In addition to impaired regulatory T-cell function, possible mechanisms for the colitis include impaired macrophage bactericidal capacity35 or increased response to toll-like receptor signaling.35,36
Many of the patients in whom diarrhea/colitis developed discontinued protocol therapy because of the severity of the diarrhea. It is likely that because of the late occurrence of this phenomenon (median time to diarrhea/colitis was 9.5 months range, 3-29), the diarrhea was initially not recognized to be a toxicity of the treatment. With increased recognition of this late-onset diarrhea/colitis, prompt interruption of treatment and intervention with oral nonabsorbable steroids may allow patients to successfully reinitiate therapy sooner. Of the 27 patients who developed grade ≥3 diarrhea/colitis, dosing was interrupted or discontinued in 21, and 11 received a corticosteroid (budesonide or prednisone). Twelve of the 27 were subsequently able to maintain dosing at either 100 or 150 mg twice daily for a minimum of 120 days.
In summary, idelalisib and rituximab produced a very high ORR in previously untreated older patients with CLL/SLL. Remissions were durable and are ongoing. In the early part of the trial, the toxicity was usually mild and easily managed. Diarrhea/colitis was a late event that was not initially assumed to be drug related. Hence, other possible causes of diarrhea were explored (the presence of Clostridium difficile, for example) and idelalisib was not necessarily discontinued. An earlier recognition of this side effect, initiation of appropriate supportive care, and early interruption of drug will likely lead to this toxicity being less severe. A phase 3 study evaluating frontline idelalisib in combination with obinutuzumab (ClinicalTrials.gov identifier NCT01980875) is ongoing and another comparing bendamustine/rituximab plus idelalisib vs placebo (NCT01980888) in previously untreated patients has completed enrollment.
Authorship
Contribution: D.M.J., L.H., L.L.M., and A.S.Y. designed the trial; S.M.O., N.L., T.J.K., I.F., A.D.Z., J.A.B., M.K., S.M., and S.E.C. performed the research; R.L.D., Y.K., and R.D.D. analyzed the data; and S.M.O., L.L.M., S.E.C., R.L.D., Y.K., and R.D.D. wrote and/or edited the manuscript.
Conflict-of-interest disclosure: S.M.O., N.L., T.J.K., I.F., A.D.Z., J.A.B., and S.E.C received institutional research grants from Gilead Sciences. L.H., A.S.Y., D.M.J., L.L.M., Y.K., R.D.D., and R.L.D. are current or former employees of Gilead Sciences.
Correspondence: Susan M. O’Brien, UC Irvine Health Chao Family Comprehensive Cancer Center, Bldg 23, 101 The City Dr S, Orange, CA 92868; e-mail: obrien{at}uci.edu.
Acknowledgments
The authors thank the patients for their dedication to this clinical trial and the clinical personnel at each of the study sites for their diligence in caring for patients and collecting study data. Timothy DiChiara, PhD, Medical Writer at Gilead Sciences, assisted in the preparation of the manuscript.
This study was sponsored by Gilead Sciences, Inc (Foster City, CA and Seattle, WA).


Hello. I have checked your osvilt.com and i see you’ve got some duplicate content so probably it is the reason that you don’t rank high in google. But you can fix this issue fast.
There is a tool that generates content like human, just search in google: miftolo’s tools