The spectrum of somatic alterations in hematologic malignancies includes substitutions, insertions/deletions (indels), copy number alterations (CNAs), and a wide range of gene fusions; no current clinically available single assay captures the different types of alterations.
We developed a novel next-generation sequencing-based assay to identify all classes of genomic alterations using archived formalin-fixed paraffin-embedded blood and bone marrow samples with high accuracy in a clinically relevant time frame, which is performed in our Clinical Laboratory Improvement Amendments–certified College of American Pathologists–accredited laboratory.
Targeted capture of DNA/RNA and next-generation sequencing reliably identifies substitutions, indels, CNAs, and gene fusions, with similar accuracy to lower-throughput assays that focus on specific genes and types of genomic alterations. Profiling of 3696 samples identified recurrent somatic alterations that impact diagnosis, prognosis, and therapy selection.
This comprehensive genomic profiling approach has proved effective in detecting all types of genomic alterations, including fusion transcripts, which increases the ability to identify clinically relevant genomic alterations with therapeutic relevance.
Introduction
In the last decade, our understanding of the somatic cancer genome has been greatly advanced through gene discovery studies.1-6 These studies delineated the genomic complexity in different types of human cancer, in different patients with the same tumor type, and within an individual’s tumor. These efforts have identified recurrent somatic alterations that in some cases are specific to a particular tumor type but in many cases are shared across different malignancies. These discoveries have had clinical impact in the diagnosis of specific malignancies defined by recurrent somatic alterations3 and in the development of more precise prognostic schema.1 Most importantly, these studies have identified disease alleles that have guided the use of molecularly targeted therapy.2 These data underscore the importance of genomic profiling in clinical oncology and led to the development of DNA-based genomic tests for cancer patients.7
Cytogenetic studies identified recurrent chromosomal translocations in a spectrum of hematologic malignancies, including acute myeloid leukemia (AML),8 acute lymphoblastic leukemia (ALL),9 chronic myeloid leukemia (CML),10 and non-Hodgkin lymphomas (NHLs),11 which impact clinical outcome and can guide therapeutic decisions.12 More recent studies have identified recurrent mutations and amplifications/deletions in a spectrum of hematologic malignancies, including AML,13 ALL, multiple myeloma (MM), and lymphoma, creating a pressing need to develop comprehensive genomic assays to identify somatic alterations in hematologic malignancies.
Hematologic malignancies have a high frequency of rearrangements that lead to aberrant expression of oncogenes or to the expression of fusion transcripts that contribute to malignant transformation and maintenance. These include classical chromosomal translocations, inversions/duplications, interstitial deletions, and episomal fusions/amplifications that can give rise to disordered expression of full-length genes or of fusion transcripts. In many cases, these discoveries have led to the use of targeted therapies in specific disease subsets.
However, current diagnostic assays, including fluorescence in situ hybridization (FISH) and real-time polymerase chain reaction (PCR), are designed ad hoc to identify specific genomic alterations, and in some cases, there are no assays that can reliably identify specific rearrangements. We sought to develop an integrated DNA/RNA profiling platform using targeted next-generation sequencing (NGS) that could reliably identify single nucleotide substitutions, insertions/deletions, copy number alterations (CNAs), and rearrangements.
Methods
Description of workflow
DNA and RNA from each patient are extracted and made into barcoded libraries through separate workflow streams. The DNA and cDNA undergo library construction and hybrid selection on independent plates. DNA and RNA samples from the same patient then converge in an analysis pipeline using the plate names and shared specimen ID. Detailed protocols for DNA and RNA extraction, cDNA synthesis, library construction, and hybrid selection are described in supplemental Information, available on the Blood Web site.
Sequencing
The selected libraries are pooled and sequenced on the Illumina HiSeq2500 to ∼500× unique coverage for DNA and to >3 M unique on-target pairs for cDNA.
Base substitutions, indels, and copy number analysis
Samples with median exon coverage in the range 150 to 250× are considered qualified, whereas those with coverage <150× are considered failed. For base substitutions, the mutant allele frequency (MAF) cutoff is 1% for known somatic variants (based on COSMIC v62) and 5% for novel somatic variants. For indels, the MAF cutoff is 3% for known somatic variants and 10% for novel somatic variants. Additional details of the methods were described previously.7
Rearrangement calling methods
A customized alignment workflow was developed for fusion detection from RNA-seq. Raw sequence reads were aligned to whole transcriptome (refSeq) first, and reads with suboptimal mapping were aligned to whole genome references. Alignments to the 2 different references were then merged and calibrated based on the full genome reference (hg19) for fusion detection.
Gene rearrangements were detected by identifying clusters of chimeric read pairs from both DNA (pairs mapping >10 kbp apart or on different chromosomes) and RNA (pairs mapping to refSeq sequences corresponding to different genes or to genomic loci >10 kbp apart). Chimera clusters were filtered for repetitive sequence (average mapq >30) and by distribution of mapped positions (SD >10). Identified rearrangements were then annotated according to the genomic loci of both clusters and categorized as gene fusions (eg, BCR-ABL1), gene rearrangements (eg, IGH-BCL2), or truncating events (eg, TP53 rearrangement). Rearrangement candidates were then filtered based on number of chimera reads supporting the rearrangement events (for documented fusions, a minimum 10 chimera reads are required; for putative somatic driver rearrangements, 50 chimera reads are required).
In addition to the de novo rearrangement detection method described above, reads were also separately aligned to a custom reference library generated based on common fusions and rearrangements. Fusions were detected based on the observation of reads aligned across the junction of rearrangement breakpoints. This method includes the detection of 6 common isoforms of MLL-PTD14 (supplemental Table 3a).
Immunoglobulin heavy locus (IGH) rearrangements were detected by targeting rearrangement hotspots of both common immunoglobulin fusion partner genes (major and minor translocations involving MYC, BCL2, and CCND1), as well as IGH breakpoint regions.
Expression measurement
We first calculated read counts per million for each gene, and then relative expression level was normalized by the median read counts per million of all 449 MM patients. The statistical significance of expression between the rearrangement positive cohort and rearrangement negative cohort was determined by a 1-sided Wilcoxon rank-sum test.
Results
NGS-based test for detecting genomic alterations in DNA and RNA
The assay we developed uses next-generation DNA and RNA sequencing and builds on the workflow that has been optimized for genomic profiling of DNA from patients with solid tumors.7 Genomic profiling is accomplished by integration of data from targeted DNA and RNA profiling with up-to-date interpretation of the clinical significance, thereby achieving increased breadth and improved sensitivity to identify rearrangements that result in aberrant expression of fusion transcripts. The genes targeted by DNAseq and RNAseq are listed in supplemental Table 1a-b, and the intervals of each exons/introns targeted by this assay are listed in supplemental Table 1c-d.
The clinical implications of targeted genes in ALL, AML, myelodysplastic syndromes (MDS)/myeloproliferative neoplasms, NHL, and MM are annotated in supplemental Table 1e. Uniform coverage across all genes are observed from a total of 108 Hapmap and hematologic tumor specimens. The distribution of coverage of 405 genes targeted in DNAseq is shown in Figure 1B, and coverage of each gene is listed in supplemental Table 1f. The distribution of exonic coverage (1244 exons) of all clinical relevant genes is shown in supplemental Figure 1. Eight exons had consistent low coverage <100× and were excluded from further analysis (supplemental Table 1a). The read depth of each gene targeted in RNAseq from 61 validation samples is listed in supplemental Table 1g. The current workflow is shown in Figure 1A and described in the supplemental Methods. The coverage of each target of genes involved in therapy, clinical trial, and/or other prognostic and standard of care is summarized in supplemental Table 1h.

Figure 1
Workflow of comprehensive combined DNA and RNA genomic analysis in clinical specimens and coverage distribution of all exons. (A) DNA and RNA are extracted from fresh blood and bone marrow aspirate (BMA) specimens procured in EDTA or FFPE biopsy/surgical specimens; cDNA from 300 to 500 ng RNA and 50 to 200 ng DNA undergoes whole-genome shotgun library construction. cDNA libraries are hybrid capture selected for 265 genes known to be rearranged in RNA and DNA libraries are hybrid capture selected for 405 genes known to be altered in DNA. Hybrid-capture selected libraries are sequenced to high depth using the Illumina HiSeq2500 platform; sequence data are processed using a customized analysis pipeline designed to accurately detect multiple classes of genomic alterations: base substitutions, short insertions/deletions, CNAs, and gene fusions; detected mutations are annotated according to clinical significance and reported. (B) Coverage distribution in DNAseq of all genes in 108 validation samples including 4 Hapmap controls and 104 hematologic tumor specimens.
Detection of DNA sequence variants and CNAs
We first established analytic accuracy of detecting substitutions, shorts insertions and deletions (indels), and CNAs by comparing the performance of the new assay with a DNA-only assay that had previously undergone comprehensive validation across a large number of clinical samples. Compared with the previously validated assay, the new assay contained an additional 90 genes relevant to hematologic malignancies. Substitution, indel, and copy number detection were validated by reanalyzing 47 samples previously profiled with a validated test7 in which 169 alterations were identified in 55 genes common to both assays (102 substitutions, 59 indels, and 8 CNAs; 10 low-frequency subclonal variants were excluded from the analysis). The concordance between the 2 sets of results was 99.4%: 168 of 169 variants were concordant with a single discordant low-frequency variant (supplemental Table 2a). Additionally, MAF values measured using the 2 assays were highly correlated (R2 = 0.929; Figure 2A). These data demonstrate that the current DNA profiling platform can accurately detect DNA sequence variants and CNAs, as established for the reference assay.
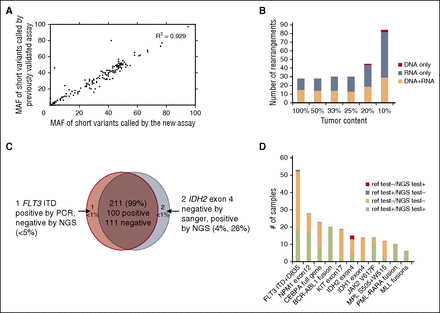
Figure 2
Alteration calling accuracy and concordance with reference platforms. (A) Correlation of short variant MAFs for substitutions and indels called by new vs previously validated assays. MAFs of 102 substitutions and 59 indels were compared in pairs and showed significant correlation (R2 = 0.929). (B) Summary of sensitivity of fusion calling in cell line mixes, separated by detection method (DNA, RNA, or both). (C) High concordance (99%) observed between NGS and Sanger sequencing or PCR validation results. (D) Concordance of alterations between the new assay and other Clinical Laboratory Improvement Amendments (CLIA)–certified reference platforms. The count of positive call and negative call from reference test and our NGS-base assay from the 11 genes/regions are highlighted by different colors: red (alteration was called negative by reference test but positive by our NGS test), steel blue (alteration was called positive in reference test and negative in our NGS test), peach (alteration was called negative in both reference test and negative test), and lime green (alteration was called positive in both reference test and negative test.
Detection of genomic rearrangements
To determine our ability to detect genomic rearrangements, we pooled 21 cell lines (Figure 3A; supplemental Table 2b) with 28 known genomic rearrangements to create 39 mixtures with ratios of 10%, 20%, 25%, 33%, and 50% representing a range of clinically relevant tumor fractions. Sensitivity and specificity of the assay were determined by comparing the genomic rearrangements detected in the pooled samples relative to the constituent cell lines. The sensitivity for fusion detection at 20% to 100% tumor fraction was 100% (161 of 161), and 98% at 10% tumor fraction (84 of 86) (supplemental Table 2b). Very few false-positive calls were observed, with a positive predictive value of >98% (245 of 248, with 2 of 3 false calls being at a marginal level of detection). The combined DNA and RNA sequencing approach accurately detects a wide variety of genomic rearrangements and gene fusions.
RNA sequencing enables efficient detection of rearrangements and targets all 28 rearrangements with novel breakpoints in a large number of genes, whereas DNA sequencing allows high sensitivity for well-characterized rearrangements affecting specific introns and rearrangements that do not result in expression of a fusion transcript, including those involving promoter regions. The distribution of rearrangements called by both DNA and RNA, RNA only, and DNA only are shown in Figure 2B. RNA sequencing maintains high sensitivity in high tumor content (100% sensitivity in tumor content 25-100%); at low tumor fraction (10-20%), DNA sequencing is more sensitive in selected introns, including, for example, breakpoints involving the MLL and PDGFRA genes.
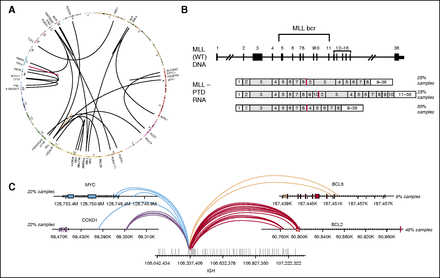
Figure 3
Combined DNA and RNA sequencing for detection of complex rearrangements. (A) Circos plot showing broad range of fusions in cell line mixes used for validation of fusion calling. (B) Three MLL-PTD isoforms that were tested (from top to bottom): duplication of MLL exons 2 to 8, exons 2 to 10, and exons 4 to 8. (C) Breakpoints on the IGH and its partner genes for the 13 clinical concordance samples including 5 IGH-MYC, 2 IGH-BCL6, 5 IGH-CCND1, and 11 IGH-BCL2 rearrangements.
Concordance with reference platforms
We performed blinded comparisons with CLIA-certified diagnostic assays, including Sequenom, reverse transcription-PCR, FISH, and PCR fragment analysis, for 76 clinical specimens (supplemental Table 2c) previously tested for 214 clinical relevant alterations in 11 genes that are known and routinely tested in clinical practice in AML, ALL, and MDS (165 short variants and 49 gene fusions; Figures 2C-D). DNA and RNA were extracted from new unstained sections from the originally tested formalin-fixed paraffin-embedded (FFPE) block. Coverage of DNA sequencing across 76 specimens is shown in supplemental Figure 2 and Table 2d. Of the 101 genomic alterations identified by Sequenom or gel sizing, 100 were also called by our new assay (1 positive FLT3 internal tandem duplicate ITD was not detected due to low mutant allele frequency).
Of 113 negative genomic alterations reported previously, our assay confirmed 111 negative calls and identified 2 IDH2 R140Q mutations that were not detected by the reference method likely due to the greater sensitivity of this approach than the orthogonal platform (Figure 2C; supplemental Table 2e). These 2 IDH2 alterations were later confirmed from an independent validation using AmpliSeq platform. Overall concordance was 99% (211 of 214; Figure 2C). The concordance between the NGS-based test and reference testing, including positive and negative calls, is shown in Figure 2D. In addition to the concordance analysis, genomic profiling of the 76 test samples identified 126 additional somatic alterations that are not covered by available hotspot assays in the given disease type, including clinically relevant genomic alterations in KRAS, TET2, EZH2, and DNMT3A (supplemental Table 2e; supplemental Figure 3).
A subset of samples with low-frequency calls (<10%) was selected for an independent validation using either AmpliSeq assay (supplemental Methods) or a CLIA-certified non-NGS mutation detection assay. Overall, 20 of 21 low-frequency variants were confirmed from AmpliSeq assay and other hotspot clinical assays (supplemental Table 2g).
Detection of complex gene rearrangements
In addition to rearrangements that result in expression of a fusion transcript containing a known or putative oncogene (Figure 3A), complex gene rearrangements are often identified in patients with hematologic malignancies. These include large intragenic rearrangements such as MLL partial tandem duplications (PTDs),15 extragenic rearrangements that involve the immunoglobulin genes, and smaller internal gene rearrangements, such as FLT3 internal tandem duplications (ITDs)16 that were assessed in the concordance with reference platforms above (Figure 2D). We profiled 12 samples that were MLL-PTD positive by PCR, spanning 3 different isoforms (Figure 3B), and 14 that were negative by PCR.
The NGS-based assay identified MLL-PTDs in 11 of the 12 PCR positive samples and confirmed the negative PCR calls in all 14 samples, for an overall accuracy of 96.5% (25 of 26, the single missed PTD was present below the calling threshold). Rearrangements involving the IGH locus commonly occur in hematologic malignancies, including NHL.17 The IGH rearrangement-calling algorithm enables detection of known IGH rearrangements in addition to novel partners. IGH rearrangement detection was assessed using 10 cell lines and 38 clinical samples harboring IGH rearrangements detected by FISH and/or karyotypic data (Figure 3C; supplemental Table 3a-b). The 10 cell lines were fully concordant, whereas the clinical samples had 2 discordant positive calls and 1 discordant negative call for an overall concordance of 94% (45 of 48) (supplemental Table 3b-c).
Assay reproducibility in clinical samples
To demonstrate assay reproducibility, we tested 13 clinical FFPE specimens in 5 replicates, each in 3 different batches with 1 batch including 3 intrabatch replicates. In total, the 65 samples contained 82 alterations (42 subs, 13 indels, 15 CNAs, and 12 rearrangements) providing a comprehensive assessment of intra- and interbatch reproducibility. Concordance between replicates was 97% for both inter- and intrabatch subsets (supplemental Table 4), with discordant calls being ascribed to sensitivity limit of the assay. Long-term reproducibility was assessed through serial assessment of 2 pooled samples (Universal Human Reference RNA for RNA, a mixture of 10 HapMap samples for DNA). These samples were sequenced repeatedly over 5 months; 303 single nucleotide polymorphisms (100% 134 of 134) in DNA and BCR-ABL fusion (100% 132 of 132) in RNA were successfully detected over the entire time series.
Workflow compatibility with different input materials
We next studied the assays performance using clinical FFPE, blood, and bone marrow aspirate samples. Blood and bone marrow were collected in EDTA for both DNA and RNA extractions. DNA and RNA were obtained from 10 different blood, bone marrow, and FFPE specimens, and nucleic acids were extracted according to protocols described in the supplemental Methods. Of these 40 samples, 39 of 40 exceeded performance specifications, with the only failing sample coming from 1 of 10 FFPE blocks analyzed (supplemental Table 5).
Clinical experience to date
The new assay was used to perform genomic profiling on 3696 hematologic malignancies submitted to our CLIA-certified, College of American Pathologists–accredited, New York State–approved laboratory; 3433 of 3696 (93%) specimens submitted for processing were successfully characterized, including 27% FFPE samples, 21% from bone marrow aspirates, 18% samples derived from blood, and 34% samples from pre-extracted nucleic acids from relevant tumor tissues. This cohort included 39% MM, 22% NHL, 17% AML, and 13% MDS/myeloproliferative neoplasm specimens (Figure 4A). Different subtypes of NHL in our clinical experiences include DLBCL (41%), follicular lymphoma (13%), B-cell lymphoma (NOS) (8%), and mantle cell lymphoma (8%) (Figure 4B). Extracted DNA was sequenced to an average depth of 500× and RNA to an average of 6.9 M unique pairs; 263 of 3696 (7%) specimens did not attain quality control specifications including failures in DNA preparation, or failures in RNA preparation, or blood sample older than acceptance criteria (1 day old) and no evidence for tumor, or no somatic driver mutations reported under qualifying conditions such as low coverage (DNA <250×), low tumor purity (<20%), and coverage bias impairing ability to call CNAs (supplemental Table 6a).
Genomic profiling of hematologic malignancies demonstrates that only a relatively small number of genes are commonly mutated, whereas many specimens harbor a wide range of rarer events (Figure 4C), including base substitutions, indels, copy number amplification/losses, and rearrangements (Figure 4D). At least 1 driver alteration was identified in 3246 of 3433 (95%) tumor specimens, and 2650 (77%) cases harbored ≥1 alteration linked to a commercially available targeted therapy or one that is in clinical development, including NRAS (14% of cases), KRAS (13%), DNMT3A (7%), CDKN2A (7%), IDH1/2 (5%), BRAF (4%), and FLT3 (4%). In addition, 61% of cases harbored ≥1 alteration with known prognostic relevance in that tumor type, including TP53 (19% of cases), ASXL1 (9%), TET2 (8%), CDKN2B (5%), CREBBP (5%), MLL (4.0%), and NPM1 (2.0%) (Figure 4C). Disease-specific mutational landscapes of MM, NHL, ALL, AML, and MDS are further presented in Figure 5 with associated clinical relevance, and a full listing of observed genes and mutation frequencies are listed in supplemental Table 6b.
Overview of clinical cases profiled by combined DNA/RNA NGS assay. (A) Tumor type distribution of all hematologic tumor cases profiled by the NGS-based DNA/RNA test. (B) Disease ontology distribution of 297 NHL tumors profiled by the assay. PMBCL, primary mediastinal (thymic) large B-cell lymphoma; AITL, angioimmunoblastic T-cell lymphoma. (C) Genomic alterations detected from the 1931 clinical specimens and distribution of alteration types. Top 50 most frequent altered genes are displayed. Red: base substitutions, steel blue: indels, peach: CNAs, lime green: rearrangement. (D) Distribution of alteration types including base substitutions, indels, CNAs, and rearrangements in the clinical specimens.
Genomic landscape in ALL, AML, MDS, NHL, and MM. Observed frequencies of most mutated genes in (A) ALL, (B) AML, (C) MDS, (D) NHL, and (E) MM. Mutation frequency is grouped by function effect, substitution/indel (red), focal amplification (steel blue), homozygous deletion (peach), rearrangement/fusion (lime green), and truncation (purple). The numbers of cases included in this analysis are 102 cases in ALL, 274 cases in AML, 130 cases in MDS, 297 cases in NHL, and 753 cases in MM. In The clinical relevance including therapeutic (green), clinical trial (olive), prognostic (lavender), and diagnostic (blue) are highlighted on top of each gene.
In total, we identified 1524 genomic rearrangements in 1256 of 3433 (37%) tumor specimens. These rearrangements involved genes implicated in chromatin/histone remodeling (20%), transcriptional regulation (16%), kinase/oncogene activation (15%), apoptosis regulation (13%), cell cycle regulation (13%), and truncation of tumor suppressors (5%), including novel in-frame fusions in kinase drug targets in ALK, BRAF, FLT3, JAK2, and ROS1 (Figure 6A). In addition, we identified a novel translocation involving NOTCH1, as well as several novel NF1 translocations that might confer sensitivity to MEK inhibitors.
Clinical highlights. (A) Novel fusions involving druggable targets such as ALK, BRAF, FLT3, JAK2, and ROS1 (5′ partner salmon, 3′ partner including kinase domain of target gene gray; ALCL, anaplastic large cell lymphoma). (B) Distribution of IGH rearrangement partners. (C) Overexpression of (i) FGFR3 and (ii) WHSC1 in t(4;14)-positive MM cases (n = 61) compared with t(4;14)-negative MM cases (n = 388). The violin plot shows the distribution of the log expression ratios of FGFR3 and WHSC1 in each group, respectively. For both cases, the difference is statistically significant (FGFR3, P = 8.7e−22; WHSC1, P = 5.28e−32, 1-sided Wilcoxon rank-sum test). (Ciii) Overexpression of CCND1 in all t(11;14)-positive cases (n = 67) compared with t(11;14)-negative cases (n = 3095). The difference is also statistically significant (P = 6.00e−38, 1-sided Wilcoxon rank-sum test). (D) Overview of short variants (single nucleotide variants SNVs, ITDs, indels) and large-scale aberrations including long deletions, copy number gains, and fusions detected in FLT3. The FLT3 protein is drawn with its immunoglobulin-like (Ig, red), transmembrane (Tm, steel blue), and tyrosine kinase (Tk, peach) domains illustrated. Positions and protein effect of all short variants are indicated. For ITDs (green), samples with ITD at the same positions are aggregated together regardless of the actual insert sequence. The ranges of large-scale aberrations are indicated with light blue bars. Commonly tested “hotspot” mutations are shaded in gray.
Rearrangements involving the IGH locus were identified in 665 of 3433 tumor specimens, including rearrangements involving CCND1/MYEOV (28%), BCL2 (25%), MMSET/FGFR3 (17%), MYC (6%), and BCL6 (4%) (Figure 6B; supplemental Table 6c). Novel IGH rearrangements were observed in 17% of cases. The locations of the most common IGH rearrangement partners are summarized in supplemental Figure 4. In addition, a sizable fraction of IGH rearrangements involved breakpoints in intergenic regions with no known oncogene within 7 MB of the breakpoint. Genomic rearrangements involving IGH are believed to lead to overexpression of the partner oncogene by placing it under the regulatory control of the IGH locus.18 Expression levels of MMSET/FGFR3 (4p16.3) and CCND1 genes were successfully measured for 449 MM cases (Figure 6C). As depicted in Figure 6C, we observed overexpression of both CCND1/FGFR3 and CCND1 genes in samples harboring the respective rearrangements.
Identification of different classes of alterations in a single gene using an integrated genomic profiling assay
Previous studies have shown that a subset of oncogenes can be activated by different types of genomic alterations in different patients.19 Constitutive activation of the FLT3 receptor tyrosine kinase can occur in a significant proportion of leukemia patients as a result of somatic ITDs, point mutations (D835/I836), or less common chromosomal rearrangements.16,20 Somatic alterations involving the FLT3 locus have been shown to be of prognostic importance in acute leukemia, and there are potent, specific FLT3 kinase inhibitors in late-stage clinical trials.21 We observed 89 alterations targeting FLT3 in 84 specimens. This included 67 (75%) D835 and ITD mutations detected by existing clinical tests. We also identified other base substitutions (n = 14), in-frame deletion (n = 1), CNAs (n = 6), and fusions involving FLT3 (n = 1) (Figure 6D). For example, 1 AML case that tested negative for D835Y harbored an I836 del, which has previously been associated with poor disease-free survival.22 Four cases positive for FLT3 ITD or D835 were found to also harbor concurrent low-frequency V592A and S451F mutations, which have been functionally validated as activating the FLT3 kinase.23 These results indicate that the sequencing-based assay can robustly detect a wide range of alterations in driver genes that are not fully evaluated using conventional methods.
Use of a single unified test in B-cell ALL
Recent genomic studies in B-cell ALL have led to an increased understanding of the molecular pathogenesis of this B-cell malignancy. This includes the identification of recurrent substitutions, indels, and fusion genes that target B-cell differentiation. Most importantly, recent studies have shown that subsets of patients with high-risk, BCR-ABL–negative B-cell acute lymphoblastic leukemia (B-ALL) have a Philadelphia chromosome-like (Ph-like) gene expression signature and that the majority of cases of Ph-like ALL harbor somatic alterations in known signaling effectors, including in known and putative therapeutic targets.24-26 However, current approaches to identify the substitution mutations, indels, and fusion genes that drive Ph-like ALL require the use of a series of genomic assays. We performed integrated DNA/RNA profiling on 16 cases classified as Ph-like ALL by gene expression profiling. We found deletions in IKZF1 (7 of 16), PAX5 (2 of 16), and CDKN2A (4 of 16) in a subset of these cases, consistent with previous reports.
We also identified known activating mutations in CRLF2, KRAS, and JAK2 in some cases. Notably, we identified known and novel gene fusions in 9 of 16 cases. This included 2 JAK2 fusions with different partner genes (PAX5 and SSBP2), 6 known fusions of CRLF2 and EPOR, and a novel gene fusion involving ABL2, all of which would be expected to activate kinase signaling and to confer sensitivity to approved drugs. In 1 case of B-ALL, we identified a FIP1L1-PDGRA fusion, which has been shown to respond to imatinib in patients with hypereosinophilic syndrome28 (Table 1). These data show that an integrated DNA/RNA profiling platform can identify a spectrum of alterations in kinase signaling pathway in B-ALL patients that can guide the use of molecularly targeted therapies. The result also achieved high concordance with an integrated research profiling approach which included genome sequencing, targeted DNA sequencing, RNA sequencing, and single nucleotide polymorphism array analysis done on the same samples. Specifically, we observed complete concordance between our clinical genomic profiling assay and research-grade genomic analysis, with the exception a CRLF2 mutation and a CDKN2A/B deletion that were detected only by the clinical sequencing assay.
Summary of genomic alterations identified in Ph-like B-ALL cases
Discussion
The identification of somatic genomic alterations with clinical relevance has increased the need for robust genomic profiling assays that can identify the different types of genomic alterations in clinically relevant cancer genes. Here we report the development of an integrated DNA/RNA target capture NGS assay, which we optimized to detect different classes of genomic events, including base substitutions, indels, CNAs, and chromosomal rearrangements. This allows the use of a single test to systematically profile clinical samples from patients with hematologic malignancies for actionable genomic lesions, in a clinically relevant time frame and quality standard. As described in results, 93% specimens were successfully profiled in the clinical setting. Seven percent of patients received failed report due to failures in DNA or RNA preparation, process of aged liquid specimen, and no somatic mutations detected under other qualifying conditions. One of the most common qualifying conditions is RNA preparation failure (132 cases), which highlighted the technical challenges of process and sequence RNA samples (supplemental Table 6a).
Previously described assays largely focused on DNA-based approaches or used gene-specific RNA tests to detect fusion transcripts in a small set of genes. We show that targeted RNA capture sequencing can be used to identify a wide spectrum of fusion genes with known roles in malignant transformation and therapeutic response and to identify novel fusions with likely diagnostic and therapeutic relevance. This approach can identify fusion transcripts present in as few as 10% to 20% of cells and can be used to show that chromosomal rearrangements result in expression of in-frame, fusion transcripts. Moreover, combining DNA and RNA capture allows for the identification of full-length transcripts driven by promoter rearrangements by DNA sequencing, as well as fusion transcripts that are more easily identified by RNA profiling. This approach has immediate clinical value in hematologic malignancies, and we believe will show significant clinical value in epithelial tumors. Of note, this approach can also be used to reliably measure expression of genes in the target set, which will inform development and testing of genomic predictors that combine mutational data and gene expression/epigenetic data to inform prognosis and to identify novel therapeutic targets.
The clinical relevance of our DNA/RNA profiling5 approach is underscored by our ability to identify genetic lesions with prognostic and therapeutic relevance in specific diseases. In the case of B-cell ALL, recent studies have identified novel genetic markers with prognostic relevance and identified novel therapeutic targets in patients with high-risk disease.24-27,29,30 The challenge has been that the critical genes implicated in B-ALL can be altered by whole gene/intragenic deletions, DNA base pair substitutions, and larger indels, as well as chromosomal, intergenic, and cryptic rearrangements that lead to expression of fusion transcripts. Currently, most centers use an amalgam of DNA, FISH, and gene-specific RNA approaches to identify a subset of the most critical genetic lesions in B-ALL. Our assay provides a single profiling platform that can reliably identify all known actionable disease alleles relevant to B-ALL to improve diagnosis and risk-adapted therapy for B-ALL patients.
Most importantly, our profiling platform, allows for the increased use of genomic profiling to improve the diagnosis and treatment of cancer patients and to more precisely match patients with targeted therapies. These efforts are critical to more broadly expand precision medicine by assuring that cancer patients can be offered genomic profiling regardless of whether they are at tertiary care centers or receive care in other settings.