
Regulation of integrins is critical for lymphocyte adhesion to endothelium and trafficking through secondary lymphoid organs. Inside-out signaling to integrins is mediated by the small GTPase Rap1. Two effectors of Rap1 regulate integrins, RapL and Rap1 interacting adaptor molecule (RIAM).
Using mice conditionally deficient in both Rap1a and Rap1b and mice null for RIAM, we show that the Rap1/RIAM module is not required for T- or B-cell development but is essential for efficient adhesion to intercellular adhesion molecule (ICAM) 1 and vascular cell adhesion molecule (VCAM) 1 and for proper trafficking of lymphocytes to secondary lymphoid organs.
Interestingly, in RIAM-deficient mice, whereas peripheral lymph nodes (pLNs) were depleted of both B and T cells and recirculating B cells were diminished in the bone barrow (BM), the spleen was hypercellular, albeit with a relative deficiency of marginal zone B cells.
The abnormality in lymphocyte trafficking was accompanied by defective humoral immunity to T-cell–dependent antigens. Platelet function was intact in RIAM-deficient animals. These in vivo results confirm a role for RIAM in the regulation of some, but not all, leukocyte integrins and suggest that RIAM-regulated integrin activation is required for trafficking of lymphocytes from blood into pLNs and BM, where relatively high shear forces exist in high endothelial venules and sinusoids, respectively.
Introduction
Regulated adhesion of lymphocytes to vascular endothelium, antigen-presenting cells, and target cells is critical for adaptive immunity.1 Among the molecules that mediate cell–cell adhesion of lymphocytes are integrins, including lymphocyte function-associated antigen 1 (LFA-1 or αLβ2) and very late antigen 4 (VLA-4 or α4β1). Integrins are cell surface receptors composed of αβ heterodimers of type I transmembrane proteins in which the extracellular domains comprise adhesion modules that interact with extracellular matrix or cognate ligands on cells. LFA-1 binds to a family of intercellular adhesion molecules (ICAM-1, ICAM-2, ICAM-3), and VLA-4 binds to vascular cell adhesion molecule 1 (VCAM-1).1 Similar to all integrins, the adhesive state of LFA-1 and VLA-4 is regulated by a process known as inside-out signaling, whereby activation events in the cell are transmitted retrograde through the receptor to modulate the conformation of the ectodomain, and thereby the affinity for its cognate ligand.2
The molecular mechanisms of inside-out signaling through LFA-1 have been intensely studied. Among the proteins that regulate inside-out signaling is the small GTPase Rap1.3 Rap1 becomes activated by one of several guanine nucleotide exchange factors, including C3G, downstream of lymphocyte activation through the antigen receptor or through chemokine receptors. Guanosine triphosphate (GTP)-bound, active Rap1 binds to several effectors. Two Rap1 effectors have been implicated in inside-out signaling to integrins, RapL4 and Rap1 interacting adaptor molecule (RIAM).5 The N-terminus of RIAM binds talin1, a cytoskeletal protein known to interact with integrins and regulate their adhesive state.2,6 A domain of RIAM that is both a pleckstrin homology and Ras binding domain targets the molecule to the inner leaflet of the plasma membrane of activated lymphocytes by functioning as a proximity detector for GTP-bound Rap1 and phosphoinositol 4,5 bisphosphate PtdIns(4,5)P2.7 Once brought to the plasma membrane, RIAM links active Rap1 to integrins via talin1.8
Whereas the immunological phenotype of LFA-1 deficiency in mice has been extensively characterized by both neutralizing antibodies9 and genetically engineered mice,10-12 the consequences of in vivo disruption of the Rap1/RIAM module has not been explored. Here we report that the Rap1/RIAM module is not required for T- or B-cell development or platelet function but is essential for efficient adhesion to ICAM-1 and VCAM-1 and for proper trafficking of lymphocytes to secondary lymphoid organs.
Materials and methods
Mice
Rap1afl/fl;Rap1bfl/fl mice were provided by Alexei Morozov (Virginia Tech Carillion Research Institute) and genotyped as described.13 RIAM−/− mice were generated by targeting embryonic stem cells (ES cells) for homologous recombination with the vector depicted in Figure 2A, such that exons 3 and 4 and an inserted neomycin resistance cassette were flanked by loxP sites. A diphtheria toxin A gene provided negative selection. Targeted ES cells were injected into blastocysts and mouse strains with germ line transmission selected. Mice null for RIAM were generated by crossing these conditional animals with B6.C-Tg(CMV-cre)1Cgn/j “Cre deleter” mice from the Jackson Laboratory. RIAM−/− mice and wild-type littermates were generated by breading RIAM+/− mice and genotyped by polymerase chain reaction, using the following primers: F1 (GAT GAG CTG TGC TGT ATG GCA CTG) + R1 (GGT AAA AAC TGT TCC TAG AAG CCA TGC) for wild-type alleles and F2 (GAA TAT CAG GAC CAGGAA TGG GAG TG) + R1 for knockout alleles.
T-cell and B-cell purification
Naive splenic T and B cells were isolated by negative selection. Briefly, spleens from wild-type and RIAM-deficient mice were harvested and single-cell suspensions were made by passing tissues through 40-μm mesh. Red blood cells were lysed in hypotonic buffer (Lonza). T cells were isolated by removing all other cells, using Dynabeads Untouched Mouse T Cells Kit (Life Technologies). B cells were isolated by removing all other cells, using Dynabeads Mouse CD43 Untouched B Cells (Life Technologies).
Adhesion assay and chemotaxis
Wells of 96-well plates were coated with recombinant 6xHis-ICAM1 and blocked with 0.5% bovine serum albumin in phosphate-buffered saline. Purified splenic T or B cells were labeled with carboxyfluorescein diacetate succinimidyl ester (Molecular Probes); suspended in phosphate-buffered saline containing 0.5% bovine serum albumin, 1 mM MgCl2, and 2 mM CaCl2; and transferred to coated plates in the absence or presence of anti-CD3 (2.5 μg/mL), anti-immunoglobulin M (IgM) (10 μg/mL), stromal cell-derived factor 1 (SDF-1) (100 ng/mL), or CXC chemokine ligand 13 (100 ng/mL) and incubated at 37°C for 30 min.
Nonadherent cell were removed by 4 consecutive washes with 0.5% bovine serum albumin-phosphate-buffered saline. Input and bound cells were measured in the 96-well plates using a fluorescence multiwell plate reader (EnVision Multilabel Plate Reader; PerkinElmer). Adhesion under flow was performed using CHO cells expressing human ICAM-1 that were grown to confluence inside ibi-treated µ-slide VI chambers (ibidi GmbH) overnight. Murine T cells were washed, labeled with carboxyfluorescein diacetate succinimidyl ester, and resuspended in serum-free RPMI. T cells ± SDF-1 (100 ng/mL) were then added and allowed to settle within the chamber for 5 minutes before beginning flow. A shear force of 1 dyn/cm2 was generated with a syringe pump (New Era Pump Systems Inc.).
Images of 5 fields per condition were acquired with an inverted Zeiss 700 laser scanning confocal microscope (Carl Zeiss Microimaging) before and after 5 minutes of continuous flow, and percentage adhesion was calculated as (postflow cell count/preflow cell count) × 100. For chemotaxis, 5 × 105 B cells in 100 µL RPMI 1640 medium containing 0.25% bovine serum albumin were added to the top chamber of 5-μm pore size filter Transwell inserts (Costar). Filters were transferred to wells containing RPMI with or without 100 ng/mL SDF-1 and incubated for 2 hours at 37°C in 5% CO2. Cells in the lower chamber were collected and counted by flow cytometry gating on lymphocytes.
Lymphocyte homing
Homing of lymphocytes to secondary lymphoid organs was measured by adoptive transfer, as described.14 Splenocytes from wild-type and RIAM-deficient mice were labeled with CMTMR (5-(and-6)-(((4-chloromethyl)benzoyl)amino)tetramethylrhodamine) or carboxyfluorescein diacetate succinimidyl ester (Molecular Probes), respectively. Equal numbers of labeled cells (1 × 107 for each) were injected into retroorbital venous sinus of RIAM−/− or wild-type mice. After 3 hours, lymph nodes, spleen, bone marrow, and peripheral blood were harvested and cell suspensions were analyzed by flow cytometry.
Antibodies
Immunoblots of cell lysates were analyzed with the following primary antibodies: rabbit anti-Rap1 (Cell Signaling Technology), mouse anti-β-tubulin (E7; developed by M. Klymkowsky and obtained from the Developmental Studies Hybridoma Bank), rabbit anti-RIAM (Abcam), and rabbit anti-RhoGDI (Santa Cruz Biotechnology Inc.). Secondary antibodies were IRDye 680-conjugated goat anti-rabbit or IRDye 800-conjugated goat anti-mouse (Li-COR). Immunoblots were visualized and quantified with the Odyssey Infrared Imaging System (Li-COR).
Additional methods can be found in the supplemental Materials available on the Blood Web site.
Results
Rap1 deficiency in T cells impairs adhesion, but not development
Two genes encode 2 isoforms of Rap1, RAP1A and RAP1B. Each isoform is ubiquitously expressed, and the functions of the 2 small GTPases appear to be redundant because mice lacking either are viable,15,16 whereas a double knockout is embryonic lethal.16 To study the effects of complete Rap1 deficiency in T lymphocytes, we crossed mice with conditional (floxed) alleles at both the RAP1A and RAP1B loci,13 with animals transgenic for Cre recombinase expressed from the Lck promoter that is activated very early in T-cell development.
No Rap1 protein was detected in T cells from Rap1afl/fl;Rap1bfl/fl;Lck-Cre mice (Figure 1A), yet T-cell development was normal (Figure 1B), indicating that Rap1 is not required for T-cell differentiation. Adhesion to ICAM-1 stimulated by engagement of the T-cell receptor or phorbol myristate acetate was markedly reduced in Rap1-deficient cells with haploinsufficient cells showing an intermediate response (Figure 1C), suggesting the levels of Rap1 are limiting in inside-out signaling in T cells. Basal adhesion was also reduced in Rap1-deficient T cells. Because T-cell trafficking through secondary lymphoid organs requires adhesion to endothelium, we studied the effect of Rap1 deficiency on T-cell populations. Whereas Rap1 deficiency did not affect the number of T cells in the spleen or thymus, there was a marked decrease in T cells in peripheral lymph nodes (pLNs) and an increase in T cells in the blood (Figure 1D). Thus, Rap1 is required for normal T-cell trafficking.

Figure 1
Rap1-deficient T cells develop normally but are defective in adhesion to ICAM-1 and homing to peripheral lymph nodes. (A) Immunoblot for Rap1 (antibody detects both Rap1a and Rap1b) of lysates of T cells, thymocytes, and splenocytes from mice of the indicated genotypes. β-tubulin serves as a loading control. (B) Percentages of thymocytes of animals with the indicated genotypes that were characterized by flow cytometry as CD4+, CD8+, double-positive, or double-negative. (C) Percentage of resting or stimulated (anti-CD3 or phorbol myristate acetate) splenic T cells from animals with the indicated genotypes adherent to ICAM-1 coated wells after washing. (D) Absolute number of CD3+ cells in thymus, 1 mL blood, spleen, and pLNs of animals with the indicated genotypes. (B, C, and D) Results plotted are mean ± SD; n = 4; **P <.01, ***P <.001.
RIAM-deficient mice are viable and fertile
To confirm the role of RIAM as an effector of Rap1 in lymphocytes, we sought to determine whether RIAM deficiency would give a phenotype in vivo similar to Rap1 deficiency. We therefore generated RIAM-deficient mice using standard methods of gene targeting in ES cells by homologous recombination (Figure 2A). In heterozygous crosses, RIAM null mice were born in Mendelian ratios, manifest no overt phenotype, and were fertile. This result suggests that other gene products can compensate for RIAM deficiency. Indeed, RIAM is a member of the MRL family of adaptor proteins that includes lamellipodin.17 Both B and T cells from RIAM−/− animals contained no immunodetectable RIAM (Figure 2B). Interestingly, neither splenocytes nor platelets from wild-type mice expressed lamellipodin, and lamellipodin expression was not induced in the spleen or platelets of RIAM-deficient animals (Figure 2C).

Figure 2
Generation of RIAM−/− mice. (A) Schematic representation of RIAM targeting. The RIAM locus of murine ES cells was targeted by homologous recombination to generate an allele with loxP sites flanking exons 3 and 4. The resulting RIAM conditional mice were crossed with a Cre-deleter strain to remove exons 3 and 4, along with the Neo cassette, and thereby generate RIAM+/− mice that were crossed to produce RIAM−/− mice. (B) Immunmoblot for RIAM and RhoGDI of lysates of T and B cells isolated from spleens of RIAM+/+ and RIAM−/− mice. (C) Immunoblot for lamellipodin and RhoGDI of lysates of of MDA-MB-231 human breast cancer cells, as well as the extracts of brain, spleen, and platelets from mice with the indicated genotype.
RIAM deficiency does not impair development of B and T lymphocytes
The percentage of double-negative, double-positive, CD4+ and CD8+ thymocytes was not affected by RIAM deficiency (Figure 3A-B), confirming that, as in Rap1 deficiency, T-cell development was not affected. Although the total number of B cells in the bone marrow (BM) of RIAM−/− mice was reduced by ∼50% (Figure 3C-D), the immature B-cell and pre-pro B-cell populations were unaffected, suggesting that B-cell development in BM, similar to that of T cells in thymus, is not altered. The marked reduction in recirculating B cells in BM (Figure 3D) suggests the B-cell deficiency in this compartment is a result of trafficking, rather than lymphopoiesis. This is consistent with observations made in talin1 null mice, which are deficient in integrin activation in lymphocytes,18 and supports that idea that, similar to egress of lymphocytes through high endothelial venules in pLNs, reentry into BM requires integrin-mediated, shear-resistant adhesion to the endothelium of the BM sinusoids.19

Figure 3
RIAM-deficient lymphocytes develop normally, but the MZ B cell population is reduced in RIAM-deficient mice. (A) RIAM+/+ and RIAM−/− thymocytes were stained with anti-CD4 and anti-CD8 Abs and analyzed by flow cytometry to reveal double-positive (DP), double-negative (DN), and single-positive (CD4+ or CD8+) cells. (B) Percentages of thymocyte subsets of animals with the indicated genotypes. Plotted are means ± SD; n = 4. (C) BM lymphocytes from RIAM+/+ or RIAM−/− mice were stained with anti-B220 and anti-IgM Abs (left) and analyzed by flow cytometry, depicting pre-pro (P; B220low, IgMlow), immature (I; B220high, IgMlow), and mature (M; B220high, IgMhigh) B-cell subsets or stained with anti-IgM and anti-IgD Abs (right) to quantify recirculating mature B cells. (D) Absolute numbers of B220+ B cells of the indicated subsets within equal volumes of BM suspensions; n = 4. (E, G) Splenocytes derived from RIAM+/+ and RIAM−/− mice were characterized by flow cytometry as total B cells or FO (CD23high, CD21low), or MZ (CD23low, CD21high) B cells (E) or T1 (IgMhigh, CD23low), T2 (IgMhigh, CD23high), or T3 (IgMlow, CD23high) transitional B cells (G). (F, H) Absolute numbers of the B cells characterized in E and G. Horizontal lines indicate mean ± SD of the individual animals plotted, *P <.05.
In contrast to BM, the number of B cells in the spleens of RIAM−/− mice was markedly increased (Figure 3E-F). Interestingly, the increase could be accounted for entirely by the follicular (FO) B cells, as the marginal zone (MZ) B cells were diminished by more than 50% (Figure 3E-F). This phenotype is similar to that of talin1-deficient animals.18 Because much more Rap1b than Rap1a is expressed in B cells, the B-cell compartment of Rap1b null mice has been studied to determine the role of Rap1 in B lymphocytes.20 Similar to RIAM−/− mice, Rap1b−/− mice had a marked reduction in MZ B cells.20 This suggests that retention of B cells in the MZ requires integrins.
Immature B cells from BM enter the white pulp of the spleen through the MZ, where they mature into transitional B cells before differentiation into MZ cells that are retained in this compartment or FO B cells that pass into the follicles of the white pulp.21 Three subsets of transitional B cells have been defined on the basis of surface markers: T1, T2, and T3.22 RIAM-deficient mice had twice the number of T2 cells in their spleens relative to wild-type animals (Figure 3G-H), suggesting a derangement in splenic maturation that may account for the surplus of FO cells at the expense of MZ B cells. Despite alterations in total numbers, all subsets of B cells are generated in RIAM−/− mice, demonstrating that, as is the case for T cells, B-cell maturation does not require RIAM.
RIAM deficiency impairs integrin-mediated adhesion of B and T lymphocytes
We next studied integrin-mediated adhesion in RIAM-deficient lymphocytes. The surface expression of neither LFA-1 (Figure 4A) nor VLA-4 (Figure 4B) was affected by RIAM deficiency in T or B lymphocytes. Nevertheless, both basal and stimulated adhesion of lymphocytes to ICAM-1 was diminished in cells derived from RIAM−/− mice (Figure 4C). This suggests that activation of LFA-1 in both B and T cells requires RIAM. Chemokine-stimulated adhesion of B cells was affected to a greater degree than that stimulated by cross-linking of the B-cell antigen receptor. Whereas T-cell-receptor-stimulated T-cell adhesion to VCAM-1 was diminished in RIAM-deficient cells, B-cell antigen receptor-stimulated adhesion of B cells to VCAM-1 was unaffected (Figure 4D), indicating that VLA-4 may be differentially regulated in B vs T cells. To better mimic physiologic conditions of adhesion under shear force, we studied the adhesion of T cells in a flow chamber in which T cells are applied to a monolayer of CHO cells that express ICAM-1. Under flow, the adhesion defect of RIAM−/− T cells was more pronounced (Figure 4E) than that measured in a static system (Figure 4C), consistent with a role for RIAM in the integrin-mediated adhesion required to resist shear forces. Chemotaxis of B cells up a gradient of SDF-1 was also diminished in the absence of RIAM (Figure 4F), suggesting that a migration defect accompanies diminished adhesion. A similar phenotype was reported for B cells from Rap1b−/− mice.20 Interestingly, this defect was not observed in talin1-deficient B cells,18 suggesting a role for RIAM in B-cell migration distinct from linking Rap1 to integrins via talin1.
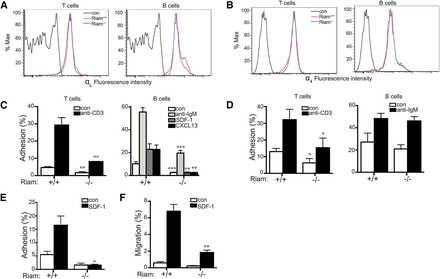
Figure 4
RIAM−/− lymphocytes are defective in integrin-dependent adhesion and migration. (A-B) Expression of LFA-1 (αL; A) and VLA-4 (α4; B) in spleen-derived T and B cells from RIAM+/+ or RIAM−/− mice was determined by flow cytometry. (C-D) Percentage of T (left) or B (right) cells (resting or stimulated as indicated) from spleens of RIAM+/+ or RIAM−/− mice adherent to ICAM-1 (C) or VCAM-1 (D) coated wells after washing. (E) T-cell adhesion to ICAM-1 ±SDF-1 measured in a flow chamber. (F) B-cell migration measured as percentage of cells that cross the membrane of a modified Boyden chamber toward media ± SDF-1. Plots show mean ± SD; n = 4 (C-D) or n = 3 (E-F). *P <.05, **P <.01, ***P <.001.
RIAM deficiency impairs homing of B and T cells to pLNs and BM, but not spleen
Given the defects in adhesion and migration of RIAM-deficient lymphocytes, we next studied trafficking to lymphoid organs. The pLNs of RIAM−/− mice were markedly smaller than those of wild-type mice (Figure 5A). The number of lymphocytes in pLNs was diminished by 85%, with both B- and T-cell compartments equally affected (Figure 5B). Histological examination of pLNs revealed dramatically shrunken B-cell zones (Figure 5C) similar to those observed in talin1-deficient animals.18 In contrast to pLNs, the spleens of RIAM−/− mice contained more B and T cells (Figure 5D), and the architecture of the white pulp was not altered (Figure 5E). More B and T lymphocytes remained in the circulation of RIAM−/− mice relative to wild-type animals (Figure 5F). These results are consistent with lymphocyte trafficking defects involving decreased entry into pLNs, but not spleen, with commensurate accumulation of B and T cells in the circulation. The entry of lymphocytes into pLNs through high endothelial venules requires cell arrest under conditions of high shear forces, a process that requires activation of integrins.23 In contrast, lymphocytes enter the marginal zone of the white pulp of the spleen from the red pulp under conditions of low flow and shear, and therefore do not require integrin activation. Thus, our observations of the derangement of lymphocyte traffic in RIAM−/− mice are perfectly consistent with a critical role for RIAM in the regulation of lymphocyte integrins.
RIAM-deficient lymphocytes traffic efficiently to spleen but not pLNs or BM. (A) Brachial, axillary, and inguinal lymph nodes removed from RIAM+/+ or RIAM−/−mice. Scale bar represents 5 mm. (B, D) Numbers of T and B cells recovered from pLNs (B) or spleens (D) of RIAM+/+ or RIAM−/−mice. Plotted are mean ± SD; n = 7; *P <.05, **P <.01. (C, E) Thin sections of inguinal and brachial pLNs (C) or spleens (E) from RIAM+/+ or RIAM−/− mice stained for B cells (B220), T cells (CD3), and stromal cells (F480). Bar represents 100 μm. (F) Numbers of T and B cells in the peripheral blood of RIAM+/+ or RIAM−/−mice. Plotted are mean ± SD; n = 4. (G) Numbers of red (CMTMR stained RIAM+/+) or green (carboxyfluorescein diacetate succinimidyl ester stained RIAM−/−) CD3+ T cells and CD19+ B cells detected by flow cytometry in the spleen, pLNs, BM, and blood of RIAM+/+ recipients 3 hours after intravenous injection of equal numbers of stained, red blood cell-depleted splenocytes. On the left is a representative cytofluorimetric scatter plot of CD19+ cells, and on the right are cumulative data plotted as the mean ± SD of the ratios of green:red cells; n = 9; ***P <.001, ****P <.0001.
To directly test the effects of RIAM deficiency on lymphocyte homing, we performed adoptive transfer of equal mixtures of fluorescently labeled lymphocytes from the spleens of RIAM+/+ (red) and RIAM−/− (green) animals and studied the ratios of red and green CD19+ or CD3+ cells that arrived 3 hours later in the spleens, pLNs, BM, and blood of wild-type recipients (Figure 5G). The ratio of green and red cells arriving in the spleen was approximately 1, demonstrating that RIAM deficiency did not affect homing to this organ. In contrast, wild-type lymphocytes homed much more efficiently to pLNs and BM than did RIAM−/− cells. The converse ratio was observed in the cells that remained in circulation, demonstrating that overall trafficking from the blood to secondary lymphoid organs was diminished by RIAM deficiency.
RIAM deficiency impairs humoral immunity to T-cell-dependent antigens
Defects in lymphocyte trafficking can affect adaptive immunity.24 To determine whether this is the case for RIAM−/− mice, we studied humoral responses to immunization. Whereas basal IgA, IgG1, and IgG3 levels were similar in RIAM+/+ and Raim−/− mice, IgM levels were significantly decreased in RIAM-deficient animals (Figure 6A). IgM and IgG3 responses to immunization with TNP-Ficoll were similar for the 2 genotypes (Figure 6B), indicating that T-cell-independent antibody responses are intact. In contrast, RIAM deficiency resulted in blunted IgM and IgG3 responses to TNP-KLH (Figure 6C), demonstrating that RIAM is required for efficient T-cell-dependent antibody production. Impaired T-cell-dependent humoral immunity was also observed in talin1-deficient mice,18 but not in Rap1b−/− animals.20 Our result is consistent with the defect in trafficking of lymphocytes to pLNs, as T-cell-dependent antibody responses require B- and T-cell interactions in these secondary lymphoid organs.25
RIAM-deficient mice are defective in T-cell-dependent humoral immunity. (A) IgA, IgM, IgG1, and IgG3 levels in serum of naive RIAM+/+ (filled circles) and RIAM−/− (open squares) mice. Data from individual animals are plotted along the Y-axis with horizontal lines indicating mean ± SD. The difference in IgM levels was statistically significant (P <.05). (B-C) Anti-TNP IgM and IgG3 levels in serum of RIAM+/+ and RIAM−/− mice measured by titration and ELISA, 7 (B-C) and 42 (C) days after intraperitoneal injection with the T-cell-independent immunogen TNP-Ficoll (B) or the T-cell-dependent immunogen TNP-KLH (C). A boost of TNP-KLH was given at day 35 (C). Titers shown are representative of 4 mice of each genotype immunized with each antigen. Anti-TNP IgM and IgG3 levels in mice immunized with TNP-KLH differed significantly at day 7 and 42 (* P <.05, ** P <.01, 2-way ANOVA).
Normal function of RIAM-deficient platelets
Because platelet function has been reported to be intact in an independently derived RIAM-deficient mouse strain,26 and because the platelet integrin, αIIbβ3, is regulated by Rap1 in a manner similar to LFA-1,27 we studied platelet function in our RIAM-deficient mice. We observed no prolonged bleeding time after tail snips in RIAM−/− mice relative to wild-type littermates, and platelets from mice with the 2 genotypes showed normal aggregation responses to Par4-activating peptide (Par4p) and the collagen mimic convulxin (Figure 7A). Using phycoerythrin-conjugated JON/A, an antibody that recognizes the activated conformation of αIIbβ3, we detected no difference in the kinetics of JON/A binding to platelets from the 2 genotypes stimulated with Par4p or convulxin (Figure 7B). Thus, whereas RIAM is required for maximal integrin-mediated adhesion of lymphocytes, it is not required for activation of the platelet integrin αIIbβ3.
Intact platelet function in RIAM−/− mice. (A) Light transmission aggregometry of washed platelets obtained from heparinized whole blood of control or RIAM−/− mice after stimulation with low-dose (LD) or high-dose (HD) Par4p (50 or 100 µM) or convulxin (75 or 150 ng/mL). Four pairs of mice were studied, and representative curves are shown. (B) Real-time activation of the αIIbβ3 integrin after stimulation with Par4p or convulxin was measured in washed platelets using JON/A-PE. Combined data from 4 pairs of mice are shown.
Discussion
Shear-resistant adhesion of lymphocytes to blood vessels is mediated by the integrins LFA-1 and VLA-4 and is critical for egress of the cells from the circulation into lymphoid tissues and sites of inflammation. To ensure that adhesion occurs at the right time and place, integrin activation is regulated by signals such as those initiated by chemokines and propagated to integrins by inside-out signaling cascades. The Ras-related small GTPase Rap1 has been shown in vitro to play a central role in the regulations of integrins.3 However, the physiological role of Rap1 in lymphocytes has not been demonstrated in vivo because of the partially redundant function of the 2 RAP1 genes, RAP1A and RAP1B, and the embryonic lethality of the double knockout.16 The analysis reported here of Rap1afl/fl;Rap1bfl/fl;Lck-Cre mice allowed the first in vivo studies of the role of Rap1 in lymphocytes. Although no lymphoid phenotype was observed in mice deficient in either Rap1a16 or Rap1b,15 our data clearly show that Rap1 is required for integrin-mediated adhesion of T lymphocytes and confirm that, in this regard, the functions of Rap1a and Rap1b are redundant. Our results demonstrate a requirement for Rap1 in T cell homing to pLNs, but not spleen. These data are consistent with those reported for LFA-1-deficient mice12 and support the idea that, whereas integrins are required for arrest of lymphocytes in high endothelial venules, a prerequisite for extravasation from the circulation into pLNs, they are not required for entry of lymphocytes into the white pulp of spleen, where shear-resistant adhesion is not required to transit the marginal sinus.21 Our data also indicate that although T-cell development in the thymus depends on the interaction of thymocytes with thymic stromal cells, this interaction does not require Rap1, and therefore may not require integrin activation.
RIAM has been characterized biochemically and in cell-based studies as a Rap1 effector involved in inside-out signaling to integrins.5,7 Silencing of RIAM in Jurkat T cells diminished adhesion.5 The results reported here are the first in vivo confirmation of this activity and the first analysis of RIAM in B lymphocytes. We show that both primary B and T cells require RIAM for integrin-mediated adhesion. We also show that RIAM is required for trafficking of both B and T lymphocytes into pLNs and BM, but not spleen, consistent with the differential requirements for integrin-mediated adhesion for egress of lymphocytes from the circulation into the various secondary lymphoid organs. Importantly, we also show that RIAM is required for a normal humoral response to a T-dependent antigen, suggesting that diminished ingress of B and T cells into pLNs and/or diminished contact of these cells with antigen-processing cells abrogates B-cell activation and differentiation into plasma cells.
The increased number of splenic B cells in RIAM−/− mice reflected an increase in follicular cells within the white pulp. Paradoxically, the number of MZ B cells was significantly decreased. This suggests that whereas integrins are required neither for B cells to pass from red to white pulp nor for maturation into follicular cells, they are required for the entrapment, maturation, and retention of these B cells in the MZ, where ICAM-1 and VCAM-1 have been shown to be expressed.28 This observation is consistent with the finding that LFA-1 and VLA-4 are required for retention of B cells in the MZ28 and with diminished MZ B cells in talin118 and Rap1b null20 mice.
Recently, another, independent RIAM−/− mouse was reported, and the authors found no defect in platelet function in these animals.26 Our results with independently generated RIAM−/− mice confirm these findings. Given the dependence of platelets on integrins, the platelet dysfunction observed in Rap1b-deficient mice,15 the robust expression of RIAM in megakaryocytes and platelets,27 and the observation that RIAM can recruit talin1 to αIIbβ3 integrins in platelets,27 the conclusion that RIAM is dispensable for platelet function is quite surprising. Because talin1 is required for αIIbβ3 activation and platelet function,29,30 and because the Rap1/RIAM complex activates talin1,6 platelets must use a distinct mechanism of talin1 activation. Because we found no lamellipodin expressed in platelets (Figure 2C), it is not clear what protein substitutes for RIAM in platelets. It is also mysterious why a distinct Rap1-dependent regulatory pathway for integrin activation evolved for platelets.
The similarity we observed between the adhesion defect of Rap1 and RIAM-deficient T cells suggests that RIAM is the principal effector of Rap1 with regard to regulating lymphocyte integrins. Because RIAM is an adapter molecule that interacts with and activates talin1,6 direct control of integrins can be explained on a biochemical basis because talin1 has been shown to also interact with the β subunit of integrins, and thereby promote dissociation of the α and β subunits, which in turn leads to a conformational change of the ectodomains of these proteins.2 However, another effector of Rap1, RapL, has also been implicated in integrin activation, and RapL-deficient mice have a phenotype remarkably similar to that reported here for RIAM−/− mice: impairment of stimulated adhesion of lymphocytes, defective trafficking to pLNs, and depleted MZ B cells in the spleen.14 Because other than a Rap1 binding domain, RIAM and RapL share no other obvious functional domains, it is difficult to conceive of how these effectors have redundant function. One feature shared by RIAM and RapL is the ability to bind to the SKAP-55/ADAP adapter molecules31,32; thus, it is possible that deficiency of either RIAM or RapL disrupt the SKAP-55/ADAP module sufficiently to block integrin activation, although how these lymphocyte adapter proteins might regulate integrins is unknown. Another possibility is that whereas RIAM may play a structural role in bringing talin1 to the plasma membrane, where it can interact with integrin β subunits, RapL may indirectly regulate signaling events critical for the maintenance of the interactions. Indeed, RapL is a splice variant of Rassf5, a member of a family of effectors that are tumor suppressors.33 RapL has been shown to interact with and regulate the kinase activity of Mst1, a Ste20 family kinase that is the mammalian ortholog of the tumor suppressor Hpo.34
The activation of RIAM through its translocation to the plasma membrane is exquisitely controlled by an AND gate constituted by simultaneous binding to GTP-bound Rap1 and PtdIns(4,5)P2.7 The mechanism whereby RapL associates with the plasma membrane has not been elucidated. The requirement for both RIAM and RapL for efficient integrin-mediated lymphocyte adhesion and trafficking represents a bifurcation downstream of Rap1 and may constitute yet another example of the fine regulation of stimulated lymphocyte adhesion that is essential to adaptive immunity but must be held in check to avoid maladaptive autoimmunity. Regardless of the interplay between the 2 Rap1 effectors relevant to integrin activation, our study reveals that the Rap1/RIAM module is essential for efficient lymphocyte adhesion and trafficking and for the humoral immune response that depends on these functions. The dispensability of RIAM for platelet function leads to the unexpected conclusion that there are at least 2 pathways by which Rap1 regulates integrins: one that depends on RIAM and one that does not.
Authorship
Contribution: F.B.G. and E.M.P. generated the RIAM−/− mice; W.S. and J.W. performed and interpreted the lymphocyte experiments; M.S. and A.M. performed the T-cell adhesion studies under flow; E.M., J.B., D.S.P., and W.B. performed the platelet studies; and W.S. and M.R.P. interpreted the data and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Mark Philips, NYU Perlemutter Cancer Institute, Smilow Research Building, Room 1205, 522 First Ave, New York, NY 10016; e-mail: mark.philips{at}nyumc.org.
Acknowledgments
The authors thank the ES cell and transgenic mouse facilities in the Koch Institute Swanson Biotechnology Center for support. The authors also thank Michael Dustin for recombinant ICAM-1 and for critical reading of this manuscript.
This work was supported by the National Institutes of Health (NIH), National Institute of General Medical Sciences (NIGMS) grants GM055279 and T32-GM007308, and National Cancer Institute (NCI) grant P30-CA016087 (M.R.P.); NIH NIGMS grant GM58801 and NCI grant U54-CA112967 (F.B.G.); and NIH National Heart, Lung, and Blood Institute grant P01-HL120846 (W.B.); Koch Institute Core Grant P30-CA14051), and funds from the Ludwig Center at MIT (F.B.G.).


