
There is no specific therapy for cardiomyopathies; therefore, all therapeutic measures are aimed at preventing complications that are incompatible with life.
Treatment of cardiomyopathy in the stable outpatient phase, with the participation of a cardiologist; periodic planned hospitalization in the cardiology department is shown to patients with severe heart failure, emergency – in cases of development of intractable paroxysms of tachycardia, ventricular extrasystoles, atrial fibrillation, thromboembolism, pulmonary edema.
Patients with cardiomyopathies need a decrease in physical activity, adherence to a diet with limited consumption of animal fats and salt, exclusion of harmful environmental factors and habits. These activities significantly reduce the load on the heart muscle and slow down the progression of heart failure.
With cardiomyopathy, it is advisable to assign diuretics to reduce pulmonary and systemic venous stasis. When violations of contractility and myocardial pumping function, cardiac glycosides are used. For the correction of heart rhythm shows the appointment of anti-arrhythmic drugs. The use of anticoagulants and antiplatelet agents in the treatment of patients with cardiomyopathies allows to prevent thromboembolic complications.
In extremely severe cases, surgical treatment of cardiomyopathy is performed: septal myotomy (resection of the hypertrophied interventricular septum) with mitral valve replacement or heart transplantation.
The definition of “cardiomyopathy” is collective for a group of idiopathic (unknown origin) myocardial diseases, which are based on dystrophic and sclerotic processes in cardiac cells – cardiomyocytes. With cardiomyopathy, ventricular function is always affected. Myocardial lesions in ischemic heart disease, hypertensive disease, vasculitis, symptomatic hypertension, diffuse connective tissue diseases, myocarditis, myocardial dystrophy and other pathological conditions (toxic, drug, alcohol) are secondary and are considered as specific secondary cardiomyopathies, morphs, medicinal, alcoholic effects) are secondary and are considered as specific secondary cardiomyopathies, morphologists, and medicinal, alcoholic effects.
Cardiomyopathy is a primary lesion of the heart muscle that is not associated with inflammatory, tumor, ischemic genesis, the typical manifestations of which are cardiomegaly, progressive heart failure and arrhythmias. There are dilated, hypertrophic, restrictive and arrhythmogenic cardiomyopathy. As part of the diagnosis of cardiomyopathy, ECG, echoCG, chest X-ray, MRI and MSC of the heart are performed. In cardiomyopathies, a sparing regimen is prescribed, drug therapy (diuretics, cardiac glycosides, antiarrhythmic drugs, anticoagulants, and antiplatelet agents); According to indications, cardiac surgery is performed.
Classification of cardiomyopathy
Anatomical and functional changes in the myocardium distinguish several types of cardiomyopathies:
- dilated (or stagnant);
- hypertrophic: asymmetrical and symmetrical; obstructive and non-obstructive;
- restrictive: obliterating and diffuse;
- arrhythmogenic right ventricular cardiomyopathy.
Causes of Cardiomyopathy
The etiology of primary cardiomyopathy today is not fully understood.
Among the likely reasons for the development of cardiomyopathy, are called:
- virus infections caused by viruses of Coxsackie, herpes simplex, influenza, etc.;
- hereditary predisposition (a genetically inherited defect causing improper formation and functioning of muscle fibers in hypertrophic cardiomyopathy);
- transferred myocarditis;
- damage to cardiomyocytes with toxins and allergens;
- endocrine regulation disorders (disastrous effect on somatotropic hormone cardiomyocytes and catecholamines);
- immune regulation disorders
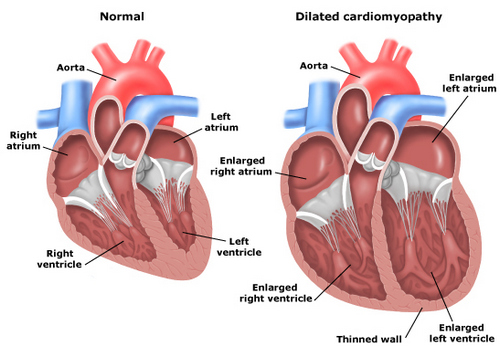
Dilatation (congestive) cardiomyopathy
Dilated cardiomyopathy (DCMP) is characterized by a significant expansion of all the cavities of the heart, symptoms of hypertrophy and a decrease in myocardial contractility. Signs of dilated cardiomyopathy manifest themselves at a young age – in 30-35 years. In the etiology of DCM, infectious and toxic effects, metabolic, hormonal, autoimmune disorders presumably play the role, in 10–20% of cases cardiomyopathy is familial in nature.
The severity of hemodynamic disorders in dilated cardiomyopathy is due to the degree of reduction in contractility and pumping function of the myocardium.
This causes an increase in pressure, first in the left and then in the right cavities of the heart. Clinically, dilated cardiomyopathy is manifested by signs of left ventricular failure (dyspnea, cyanosis, cardiac asthma and pulmonary edema), right ventricular failure (acrocyanosis, liver enlargement, ascites, edema, swelling of the veins of the neck), heart failure, unrecoverable nymphs, ascites, edema, swelling of the veins of the neck, heart failure, unrecoverable nymphs, ascites, edema, swelling of the veins of the neck, heart failure, unrecoverable nymphs, ascites, edema, swelling of the veins of the neck, heart failure, unrecoverable nymphs, ascites, edema, swelling of the veins of the neck, heart failure, unrepaired
Objectively marked deformation of the chest (heart hump); cardiomegaly with the expansion of the left, right and up; deafness of heart tones at the apex, systolic murmur (with relative insufficiency of the mitral or tricuspid valve), canter rhythm are heard. When dilated cardiomyopathy revealed hypotension and severe arrhythmias (paroxysmal tachycardia, extrasystole, atrial fibrillation, blockade).
In an electrocardiographic study, predominantly left ventricular hypertrophy and cardiac conduction and rhythm disturbances are recorded. EchoCG shows diffuse myocardial damage, sharp dilatation of the cardiac cavities and its predominance over hypertrophy, intactness of the heart valves, diastolic dysfunction of the left ventricle. Radiographically with dilated cardiomyopathy is determined by the expansion of the borders of the heart.
Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is characterized by a limited or diffuse thickening (hypertrophy) of the myocardium and a decrease in the ventricular chambers (mostly left). HCM is a hereditary pathology with an autosomal dominant type of inheritance, often develops in men of different ages.
In the hypertrophic form of cardiomyopathy, symmetric or asymmetric hypertrophy of the muscle layer of the ventricle is observed. Asymmetric hypertrophy is characterized by the predominant thickening of the interventricular septum, symmetrical hypertrophic cardiovascular disease – by a uniform thickening of the ventricular walls.
On the basis of ventricular obstruction, there are 2 forms of hypertrophic cardiomyopathy – obstructive and non-obstructive. In obstructive cardiomyopathy (subaortic stenosis), the outflow of blood from the cavity of the left ventricle is disturbed, and in non-obstructive hcmp, there is no stenosis of outflow tracts. The specific manifestations of hypertrophic cardiomyopathy are the symptoms of aortic stenosis: cardialgia, dizziness, weakness, fainting, palpitations, shortness of breath, pallor. In later periods join the phenomenon of congestive heart failure.
Percussion is determined by an increase in the heart (more to the left), auscultatory – deaf heart sounds, systolic sounds in the III-IV intercostal space and in the area of the apex, arrhythmias. Is determined by the displacement of the cardiac shock down and left, small and slow pulse at the periphery. ECG changes in hypertrophic cardiomyopathy are expressed in myocardial hypertrophy of predominantly left heart, inversion of T wave, registration of abnormal Q wave.
The most informative of non-invasive diagnostic methods for HCM is echocardiography, which reveals a decrease in the size of the cardiac cavities, thickening and poor mobility of the interventricular septum (with obstructive cardiomyopathy), a decrease in myocardial contractility, anomalous systolic mitral valve prolapse.
Restrictive cardiomyopathy
Restrictive cardiomyopathy (RCMP) is a rare myocardial lesion, usually occurring with endocardial interest (fibrosis), inadequate diastolic ventricular relaxation and impaired cardiac hemodynamics with preserved myocardial contractility and no marked hypertrophy.
In the development of RCMP, a significant role is played by pronounced eosinophilia, which has a toxic effect on cardiomyocytes. With restrictive cardiomyopathy, endocardial thickening and infiltrative, necrotic, fibrotic changes in the myocardium occur.
The development of the RCMP goes through 3 stages:
- Stage I , necrotic, is characterized by pronounced eosinophilic myocardial infiltration and the development of coronaritis and myocarditis;
- Stage II – thrombotic – manifested by endocardial hypertrophy, parietal fibrinous overlays in the cavities of the heart, vascular myocardial thrombosis;
- Stage III – fibrotic – is characterized by widespread intramural myocardial fibrosis and nonspecific obliterating endarteritis of the coronary arteries.
Restrictive cardiomyopathy can occur in two types: obliterating (with fibrosis and obliteration of the ventricular cavity) and diffuse (without obliteration). With restrictive cardiomyopathy, there are severe, rapidly progressive congestive circulatory failure: severe shortness of breath, weakness with little physical effort, increasing edema, ascites, hepatomegaly, swelling of the veins of the neck.
In size, the heart is usually not enlarged, with auscultation, the canter rhythm is heard. Atrial fibrillation, ventricular arrhythmias are recorded on the ECG, a decrease in the ST segment with T-wave inversion can be determined.
Radiological signs of venous stasis in the lungs, slightly increased or unchanged heart size. The ultrasound picture reflects the failure of the tricuspid and mitral valves, reducing the size of the obliterated ventricular cavity, impaired pumping and diastolic function of the heart. Eosinophilia is noted in the blood.
Arrhythmogenic cardiomyopathy
The development of arrhythmogenic right ventricular cardiomyopathy (AKPZH) characterizes the progressive replacement of right ventricular cardiomyocytes by fibrous or adipose tissue, accompanied by various disorders of ventricular rhythm, including ventricular fibrillation.
A rare and poorly understood disease, heredity, apoptosis, viral and chemical agents are called possible etiological factors.
Arrhythmogenic cardiomyopathy may develop as early as adolescence or adolescence and manifests as a heartbeat, paroxysmal tachycardia, dizziness, or fainting. Further development of life-threatening types of arrhythmias is dangerous: ventricular extrasystoles or tachycardias, episodes of ventricular fibrillation, atrial tachyarrhythmias, atrial fibrillation or flutter.
In arrhythmogenic cardiomyopathy, the morphometric parameters of the heart are not changed. Echocardiography renders moderate dilation of the right ventricle, dyskinesia, and local protrusion of the apex or lower wall of the heart. MRI revealed structural changes in the myocardium: local thinning of the myocardial wall, aneurysm.
Complications of cardiomyopathy
In all types of cardiomyopathy, heart failure progresses, arterial and pulmonary thromboembolism, cardiac conduction disorders, severe arrhythmias (atrial fibrillation, ventricular extrasystole, paroxysmal tachycardia), and sudden cardiac death syndrome may develop.
Diagnosis of cardiomyopathy
In the diagnosis of cardiomyopathy, the clinical picture of the disease and data from additional instrumental methods are taken into account. The ECG usually shows signs of myocardial hypertrophy, various forms of rhythm and conduction disturbances, changes in the ST segment of the ventricular complex.
When radiography of the lungs can be identified dilatation, myocardial hypertrophy, congestion in the lungs.
Especially informative for cardiomyopathy data Echocardiography, determining the dysfunction and myocardial hypertrophy, its severity and the leading pathophysiological mechanism (diastolic or systolic failure). According to the testimony, it is possible to conduct an invasive examination – ventriculography.
Modern methods of visualization of all parts of the heart are MRI of the heart and MSCT. Sounding of the cavities of the heart allows for the collection of cardio biopsies from the cavities of the heart for morphological research.
Prognosis for cardiomyopathy
Regarding the prognosis, the course of cardiomyopathy is extremely unfavorable: heart failure is steadily progressing, the probability of arrhythmic, thromboembolic complications and sudden death is high.
After the diagnosis of dilated cardiomyopathy, 5-year survival is 30%. With systematic treatment, it is possible to stabilize the condition indefinitely. Cases exceeding 10-year patient survival after heart transplantation operations are observed.
Surgical treatment of subaortic stenosis in hypertrophic cardiomyopathy, although it gives an undoubted positive result, is associated with a high risk of patient death during or shortly after surgery (every 6th operated patient dies). Women with cardiomyopathy should refrain from pregnancy due to the high probability of maternal mortality. Measures for the specific prevention of cardiomyopathy have not been developed.

Thankyou for your practical opinion on what is normally an overlooked topic. Is it okay to link this with my group?
Hi, I think your site might be having browser compatibility issues. When I look at your website in Safari, it looks fine but when opening in Internet Explorer, it has some overlapping. I just wanted to give you a quick heads up! Other then that, fantastic blog!
You’ve made some good points there. I checked on the net to find out more about the issue and found most individuals will go along with your views on this website.